[ Deutsch ] [ English ] [ Русский ]
Herzlich willkommen!
molekulartherapie.de
Eine universelle Theorie & Therapie der Erkrankungen des zentralen Nervensystems
Sonstige Noxen und Affektive Störungen
Kapitel 4 B: Sonstige exogene Noxen als Ursachen Affektiver Störungen
4.11 Kurzer Rückblick auf Kapitel 4 A und Ausblick auf Kapitel 4 B ▲
Auswirkungen potentieller Zellschwachstellen auf Affekterkrankungen
Potentielle somatische Mutationen der Zell-DNA und/oder ein potentieller Mangel bzw. Mängel an acht Kausalfaktoren ergeben neun Zellschwachstellen:
- Somatische Zell-DNA-Mutationen
- Aminosäurenmangel
- Mikronährstoffmangel
- Fettsäuren- und Fettbegleitstoffmangel
- Wassermangel
- Veränderungen der ursprünglichen Erbinformation
- Probleme mit der Glukoseverstoffwechslung bzw. -versorgung
- Sauerstoffmangel
- Probleme mit der Synthese nicht-codierender Ribonukleinsäuren
Diese neun Ereignisse können jeweils unterschiedliche Ursachen und Folgen haben:
- Hinsichtlich der Entstehung somatischer Mutationen werden (1) Informationsübertragungsfehler, induzierte Zell-DNA-Veränderungen (2) natürlicher und (3) nicht-natürlicher Art durch Noxen bzw. Mutagene und (4) physiologische Mutationen ohne erkennbare Ursachen unterschieden. Darüber hinaus spielen die (5) Weitergabe und Anhäufung von Mutationen der Zell-DNA (Fehlerakkumulation) eine bedeutende Rolle.
Mutationen betreffen Substanzen, die mittels der Zell-DNA-Codes hergestellt werden, also Peptide (insbesondere Enzyme oder Proteine) und nicht‑codierende Ribonukleinsäuren (ncRNA).
Sowohl bei der Informationsübertragung während der Zellteilung als auch bei der Informationsübertragung von der DNA auf die Boten-RNA während der Proteinbiosynthese kann es zu Veränderungen des DNA-Codes kommen.
Induzierte natürliche Zell-DNA-Mutationen entstehen durch täglichen, unvermeidbaren Zellstress, beispielsweise aufgrund von Sauerstoffradikalen, welche die Folge der lebenserhaltenden Zellatmung sind, oder aufgrund extern einwirkender Noxen, beispielsweise zellschädigender Strahlung (natürliche radioaktive Hintergrundstrahlung oder UV-Strahlung), der sich ebenfalls niemand entziehen kann.
Induzierte Zell-DNA-Mutationen nicht-natürlicher Art entstehen demgegenüber durch zusätzlichen und ‑ zumindest theoretisch ‑ vermeidbaren Zellstress, wozu beispielsweise die über die natürliche Hintergrundstrahlung hinausgehende ionisierende Strahlung gehört. Das ist zum Beispiel die radioaktive Strahlung nach der Havarie eines Atomreaktors oder aufgrund medizinischer Anwendungen.
Jede Zelle verfügt zwar über DNA-Reparaturmechanismen, dennoch können Mutationen nicht gänzlich vermieden oder beseitigt werden, und im Laufe der Zeit häufen sich somatische Mutationen an, was als Akkumulation bezeichnet wird. Das Gelingen der DNA-Reparatur hängt in großem Maße auch von der Verfügbarkeit bzw. Qualität der acht Kausalfaktoren ab: Je schlechter die Kausalfaktorenversorgung ist, desto weniger erfolgreich sind Reparaturprozesse, desto häufiger und schneller entstehen somatische DNA-Mutationen.
Somatische DNA-Mutationen haben unterschiedliche Auswirkungen. Eine als „stumm“ bezeichnete Mutation ist ohne jegliche Veränderungswirkung auf den Zellstoffwechsel, während eine nicht-stumme DNA-Mutation zu geringeren bis schwersten Zellprozessstörungen führen kann ‑ bis zum Zelltod. Die Wirkung einer DNA-Mutation hängt von der Bedeutung des geschädigten Peptid- oder ncRNA-Moleküls ab: Je bedeutender das Molekül, desto schwerer potentiell mögliche Folgen.
- Die Versorgung mit den Kausalfaktoren Aminosäuren, Mikronährstoffen, Fetten/Fettbegleitstoffen und Wasser ist von Ernährung und körperlicher Verfassung abhängig. Die allgemeine zellphysiologische Relevanz dieser Substanzen ist eher gering. Das liegt u. a. auch daran, dass es im Vergleich mit anderen Kausalfaktoren im Durchschnitt seltener zu Versorgungsproblemen kommt und diese mittels Substitution zu beheben sind.
Problematisch sind insbesondere eine zu geringe Zufuhr durch eine Mangel- oder sonstige Fehlernährung oder erkrankungsbedingte Mangelversorgung. So können Erkrankungen des Verdauungssystems zu Nährstoffaufnahmestörungen führen, Erkrankungen des Gefäßsystems führen häufig zu Problemen der Nährstoffabgabe an die Zellen.
Für spezielle Personenkreise sind ernährungsabhängige Kausalfaktoren daher bedeutender, beispielsweise für Patienten mit Erkrankungen des Verdauungssystems oder bestimmten Ernährungsweisen. So ist die Relevanz der Vitamin-B12-Versorgung für Veganer und Menschen mit Magen- oder Darmerkrankungen höher als bei der davon nicht betroffenen Bevölkerung, auch im Zusammenhang mit Affektstörungen. Ein weiteres Beispiel sind ältere Menschen, die im Schnitt zu wenig essen und trinken. Daraus ist zu schlussfolgern, dass ernährungsabhängige Kausalfaktoren auch bei ihnen eine höhere Relevanz haben als beim Rest der gut versorgten Bevölkerung. Das gilt natürlich in vergleichbarer Weise für die Bevölkerung in Weltregionen mit generell problematischer Nahrungsversorgung.
- Durch die ursprüngliche Erbinformation der Elterngeneration können DNA-Mutationen aus früheren Generationen oder aufgrund von Fehlern während der Keimzellenentstehung oder -verschmelzung in den entstehenden Organismus eingeschleppt werden. Die allgemeine zellphysiologische Relevanz der ursprünglichen Erbinformation wird als mittel eingestuft und ist damit höher als die der ernährungsabhängigen Faktoren. Veränderungen der ursprünglichen Erbinformation können zu leichteren bis schwerwiegenden Erkrankungen führen, müssen aber nicht zwangsläufig negative Folgen haben.
Drei Arten von Erbinformationsveränderungen können zu unterschiedlichen Störungen führen. Genetische Erkrankungen im eigentlichen Sinne haben diagnostisch gut erfassbare Merkmale bzw. Symptome, die meist auf einem einzigen veränderten DNA-Code beruhen und einen regelmäßigen Erbgang aufweisen. Syndrome treten nach Veränderungen der Chromosomenanzahl oder größerer Chromosomenstrukturveränderungen auf. Bei der polygenetischen Prädisposition gelten multiple Genveränderungen lediglich als Mitverursacher einer Erkrankung, die nur zusammen mit weiteren negativen Einflüssen bzw. Noxen entstehen kann.
Es wurde noch kein Hinweis gefunden, dass es sich bei Affektiven Störungen um genetisch bedingte Erkrankungen im engeren Sinne mit einem regelmäßigen Erbgang handelt. Bei bestimmten Affekterkrankungen wurden empirisch polygenetische Einflussfaktoren festgestellt. Es hat sich der Verdacht erhärtet, dass diese vor allem für die Entstehung einer Bipolaren Störung mitverantwortlich sein könnten, während die Entstehung einer unipolaren Depression weniger von polygenetischen Einflussfaktoren abhängt, der Nachweis eines Kausalzusammenhangs konnte noch nicht erbracht werden.
Die spezielle Relevanz der ursprünglichen Erbinformation könnte für die Bipolare Störung damit etwas höher sein, während sie für die Entstehung einer unipolaren Depression wahrscheinlich eher gering ist.
- Eine kontinuierliche, nicht schwankende Glukoseversorgung ist vor allem vom Zustand der Blutzuckerregulierungssysteme abhängig. Das Ernährungsverhalten folgt an zweiter Stelle. Es wurde eine im Schnitt mittlere zellphysiologische Relevanz für Glukose festgestellt, die damit höher bewertet wurde als die Versorgung mit den restlichen ernährungsabhängigen Kausalfaktoren.
Analysen und emprische Untersuchungen erhärten den Verdacht, dass die Relevanz der Glukose bei Affektiven Störungen aus drei Gründen über der allgemeinen zellphysiologischen Relevanz liegt. Die körpereigenen Blutzuckerregulationssysteme geraten nämlich (1) relativ schnell außer Kontrolle und erhalten (2) in der Breitenmedizin keine Aufmerksamkeit, so dass damit verbundene negative Auswirkungen nicht konsequent behoben werden. Dazu kommt, dass Nervenzellen (3) ausschließlich Glukose für ihre Energieversorgung verwenden.
- Eine ausreichende Versorgung mit Sauerstoff wird vor allem von einem funktionstüchtigen Atemsystem garantiert. Atemwegserkrankungen behindern die Sauerstoffaufnahme, beispielsweise das Schlafapnoë-Syndrom oder schwere Lungenerkrankungen wie COPD. Auch vaskuläre Probleme, bedingt durch ein krankhaft verändertes Gefäßsystem, können u. a. zu einem kurz- oder langfristigen Sauerstoffmangel führen.
Für das Nervensystem kommt erschwerend hinzu, dass es auf Sauerstoffmangel besonders empfindlich reagiert und es schnell zum Untergang von Nervenzellen durch die besonders gefährliche Zellnekrose kommen kann.
Die Relevanz des Sauerstoffs liegt im Allgemeinen daher im mittleren Bereich. Aufgrund seiner für das Zentralnervensystem entscheidenden Bedeutung ist eine höhere Bewertung begründbar.
- Nicht-codierende Ribonukleinsäuren (ncRNA) werden in jeder Zelle ausschließlich durch DNA-Transkription synthetisiert und haben entscheidende Einflüsse auf die Proteinbiosynthese. Ohne eine ausreichende (Selbst-)Versorgung mit ncRNA ist keine Zelle in der Lage, genügend Enzyme und Proteine herzustellen, um ihre Aufgaben korrekt erfüllen zu können.
Eine dysfunktionale ncRNA‑Synthese beruht entweder direkt auf fehlerhaften ncRNA‑Codes oder indirekt auf Problemen mit Enzymen, die für die ncRNA‑Synthese benötigt werden; letzteres könnte auch an fehlerhaften DNA-Codes der entsprechenden Enzyme (Genen) liegen. Beides entsteht durch somatische DNA‑Mutationen, die sich im Laufe der Zeit auf dem Genom anreichern („akkumulieren“, → DNA‑Mutationen oben), sie können aber auch auf Schäden der ursprünglichen Erbinformation beruhen.
Die ncRNA-Codes sind dabei in Gänze häufiger von Veränderungen oder Schäden betroffen als Gene. Der Grund ist eine deutlich höhere Anzahl von ncRNA-Codes im Vergleich zu Peptid-Genen. Aktuelle Schätzungen gehen davon aus, dass 95% des aktiven menschlichen Genoms mit ncRNA-Codes belegt ist.
Die Ergebnisse bestätigen die schon in Kapitel 3 festgestellte hohe allgemeine zellphysiologische Relevanz von ncRNA-Molekülen auch für den speziellen Fall Affektiver Störungen. Durch neueste Erkenntnisse der RNA-Forschung steigt die allgemeine Bedeutung der ncRNA weiter.
Zellprozessprobleme sind die unmittelbaren Konsequenzen dieser neun Mangelszenarien. Sie führen kurz-, mittel- oder langfristig zu einem Rückgang der Zellaktivitäten, so dass davon betroffene Organe ihre Aufgaben nicht mehr optimal ausführen können und Erkrankungen entstehen. Sind affektrelevante Gehirnregionen betroffen, können Affektstörungen die Folgen sein.
Ausblick auf die Inhalte von Kapitel 4 B
Neun Zellschwachstellen stellen vor allem mit weiteren endogenen oder exogenen Einflüssen ‑ Noxen genannt ‑ ein Gefahrenpotential für Zellen dar; die Noxen nutzen Zellschwachstellen quasi als Einfallstor für ihren negativen Einfluss auf den Zellstoffwechsel. Einige Noxen können aber auch unabhängig von Zellschwachstellen schädigen.
In den folgenden Abschnitten 4.12 ff. werden Verbindungen herausgearbeitet, die zwischen der jeweiligen Noxe und dem kausaltheoretischen Szenario (→ Kapitel 1) bestehen: Wie kann es dazu kommen, dass eine bestimmte Noxe XY die Zellen des zentralen Nervensystems so (schwer) belastet, dass es zu den im 3‑Stufen‑Modell beschriebenen Situationen in bestimmten Hirnarealen kommt, die mit verschiedenen Dysfunktionalitäten bzw. Affektstörungen verbunden sind?
Die Darstellung erfolgt systematisch mit Hilfe einer Unterscheidung dreier Hauptkategorien von Noxen:
- Psychosoziale Noxen bzw. psychosozialer Disstress
- Abiotische physikalisch-chemische Noxen, zum Beispiel Lärm, Radioaktivität oder Giftstoffe
- Biotische biologisch-medizinische Noxen, zum Beispiel Schilddrüsenerkrankungen, Viren oder Bakterien
Hinweis: Eine vollständige separate Analyse der in Teil A erörterten Zellschwachstellen getrennt von sonstigen exogenen Noxen des Teils B ist wegen der komplexen Zusammenhänge nicht möglich, ebenfalls gibt es Zusammenhänge innerhalb der verschiedenen Noxen. In solchen Fällen erfolgen entsprechende Verweise bzw. der Sachverhalt wird unter dem neuen Gesichtspunkt noch einmal erörtert, wobei es zwangsläufig zu redundanten Darstellungen kommen kann.
4.12 Psychosozialer Disstress und Affektstörungen ▲
Wie ist es möglich, dass psychisch-soziale Belastungen des Alltags in einer manifesten Erkrankung des Gehirns bzw. einer affektiven Erkrankung münden? Um diese Frage auf kausaltheoretischer Grundlage schlüssig beantworten zu können, muss zunächst geklärt werden, was unter psychisch-sozialen Belastungen überhaupt verstanden werden soll.
4.12.1 Ein psychosoziales (Dis-)Stressmodell
Leider gibt es keine einheitliche Stressdefinition oder Stresstheorie, stattdessen werden verschiedene Stressbegriffe und Stressmodelle diskutiert.
Eine einheitliche Auffassung dessen, was Stress genau ist und welche Folgen er hat, ist jedoch notwendig, um dessen Auswirkungen auf die Genese neurologisch-psychiatrischer Erkrankungen plausibel nachvollziehen zu können. Dazu muss das Rad auch nicht komplett neu erfunden werden, da mit Hilfe von Elementen und Begriffen bestehender Stresstheorien ein geeignetes Modell konstruiert werden kann.
Reaktionen auf Stress als kurzfristige Überlebensstrategien
Stressreaktionen sind überlebensnotwendig. Sie erfolgen auf einen Stressauslöser und erhöhen seit Jahrmillionen die Überlebenschance von Tier und Mensch bei gefährlichen Situationen. In der Frühgeschichte waren das meist Angriffe von Raubtieren oder anderen Menschen oder die entscheidenden Augenblicke während der Jagd. Daher sind Stressreaktionen von der Natur nur für die Zeitdauer von Sekunden oder Minuten vorgesehen.
Mit drei kurzfristigen Stressreaktionen kann sich ein Mensch rasch aus Gefahrensituationen manövrieren, indem er blitzschnell angreift, flüchtet oder in Erstarrung verharrt. Insbesondere das zentrale Nervensystem hat die Aufgabe, dafür die Voraussetzungen zu schaffen, indem es die geistig-körperliche Situation entsprechend schnell anpasst und nach beendeter Gefahrensituation wieder den Normalzustand herstellt.
Glücklicherweise treten urzeitliche Gefahrensituationen nur noch selten auf, hungrige Löwen sind in den meisten Ländern höchstens im Zoo zu sehen. Vergleichbare kurzfristige lebensbedrohliche Situationen ereignen sich in unserer Zeit beispielsweise im Straßenverkehr.
Leider ist es aber heutzutage so, dass sich zusätzlich zu akut-lebensbedrohlichen Situationen vermehrt solche ergeben, welche die gleichen körperlichen Stressreaktionen hervorrufen und Menschen kurz-, mittel- und vor allem langfristig belasten, beispielsweise die Angst um den Arbeitsplatz, Armut, Mobbing, Stalking, chronischer Leistungsdruck, sexuell-körperliche Misshandlungen, kriegerische Handlungen, Lärm oder dysfunktionale Familienverhältnisse (→ Tabellen 19 und 21). Weitere Stressfaktoren sind die vielen gesellschaftlich-sozialen Normen der modernen Zeit, die einen spontanen Frustrationsabbau unmöglich werden lassen.
Langfristige Stresssituationen gab es in der Frühzeit des Menschen wahrscheinlich auch, aber sie hatten andere Gründe und Konsequenzen als heute. So gab es mit hoher Wahrscheinlichkeit Existenzängste wegen Hungers und anhaltender Jagdmisserfolge, diese wurden aber oft nicht überlebt, da die Menschen an Entkräftung starben.
Überhaupt endeten viele ‑ auch kurzfristige ‑ frühzeitliche Stresssituationen unmittelbar mit dem Tode. So konnten sich nicht immer alle erfolgreich vor Raubtieren oder anderen lebensbedrohenden Feinden in Sicherheit bringen.
Zusätzlich muss die frühzeitliche niedrige Lebenserwartung berücksichtigt werden; schon aus diesem Grunde konnte Stress oft gar keine langfristige Folgen haben, da Menschen ein Überleben nur selten über das 30. Lebensjahr hinaus gelang.
Zwei grundsätzliche Formen von Stress: Eustress und Disstress
Beim Eustress handelt es sich um anregend wirkenden positiven Stress. Hier hat man es mit Herausforderungen zu tun, die dem Individuum Freude bereiten. Eustress entsteht beispielsweise bei (freiwilligem) Sport, auf als Bereicherung empfundenen Reisen und vor allem bei einer für positiv befundenen beruflichen Tätigkeit. Eustress fördert das psychische und körperliche Wohlbefinden, erhöht die Aufmerksamkeit und Leistungsfähigkeit. Auch lang anhaltender Eustress ist positiv, solange die körperlich-geistigen Grenzen nicht überschritten werden, andernfalls besteht die Gefahr eines Übergangs in den Disstress.
Disstress ist negativer Stress. Er entsteht, wenn man sich in einer unsicheren, unangenehmen oder bedrohlichen Situation befindet, der man entkommen will bzw. muss oder einer erheblichen körperlich-geistigen Überforderung ausgesetzt ist. Disstress gefährdet das körperliche und psychische Wohlbefinden und vermindert Aufmerksamkeit und Leistungsfähigkeit.
Auch vermeintlich positive Ereignisse können Disstressoren sein, zum Beispiel wenn mit ihnen ein hoher Erwartungsdruck oder Unsicherheitsgefühle einhergehen, wie Heirat, Familienzuwachs oder überhaupt jede grundlegende Lebensveränderung.
Die Einhaltung gesellschaftlich-religiöser Normen führt zu Disstress, wenn dies für das Individuum erhebliche Nachteile bedeutet, beispielsweise geschlechtsspezifische Unterdrückung oder die kirchliche Ächtung bzw. das Verbot von Ehescheidungen, durch das zerrüttete Ehe- und Familienverhältnisse nicht bereinigt werden können.
In den hier zu erörternden Zusammenhängen geht es ausschließlich um kurz-, mittel- und langfristigen Disstress und dessen Auswirkungen auf der körperlichen und affektiv-emotionalen Ebene und den Zusammenhängen zwischen beiden Ebenen.
Entstehung und Lösung von Disstresssituationen
Drei Phasen charakterisieren die Entstehung und Beseitigung negativen Stresses (→ Abbildung 36):
(1) Disstressauslöser → (2) Realisations- und Aktivitätsphase → (3) Disstressauflösung
In Phase 1 entsteht negativer Stress, das Individuum gerät in eine unangenehme bzw. (lebens-)bedrohliche Situation.
Die eigentliche Belastung findet in Phase 2 statt und beginnt mit der Realisation der unangenehmen bzw. bedrohlichen Situation. In diesem Zeitraum kann sich das Individuum um Stressbewältigung bemühen, indem es die Stressursache beseitigt oder sich dieser anpasst; unterbleiben diese Aktivitäten, verbleibt es im Disstress, sofern sich die Situation nicht von selber bereinigt.
Die Disstressauflösung in Phase 3 ist das Ergebnis einer erfolgreichen Aktivitätsphase durch vollständige Stressbeseitigung. Das Individuum befindet sich wieder im stressfreien Normalzustand.
Ob eine Person die Situation in ihrem Sinne aktiv lösen kann, hängt von ihrer Stressbewältigungs- bzw. Lösungskompetenz für den speziellen Disstressor ab. Verfügt jemand von sich aus nicht über eine ausreichende Lösungskompetenz, kann diese mit Hilfe externer Unterstützung erhöht werden.
Verhaltensmuster bzw. Strategien zur Disstressbewältigung
Aus den drei „prähistorischen“ Überlebensstrategien ‑ Kampf, Flucht oder Erstarrung ‑ sind unter den heutigen, veränderten Bedingungen fünf Verhaltensmuster geworden. Nur bei den ersten drei handelt es sich um sinnvolle Strategien zur Disstresslösung, die beiden letzten sind kontraproduktive Ausweichreaktionen:
- Anpassung → Sich stärken, um dem Disstressor gewachsen zu sein (indirekte Disstressor-Beseitigung)
- Veränderung → Beseitigung oder Abmildern des Disstressors (direkte Disstressor-Beseitigung)
- Akzeptanz → Den Disstressor bewusst hinnehmen und lernen, positiv damit umzugehen
- Stellvertreterkrieg → Ärger und Frustrationen verlagern, um sich vom Disstressor abzulenken
- Resignation oder die „erlernte Hilflosigkeit“ → Keine Aktivitäten
ABBILDUNG 36: ENTSTEHUNG UND BESEITIGUNG VON STRESS
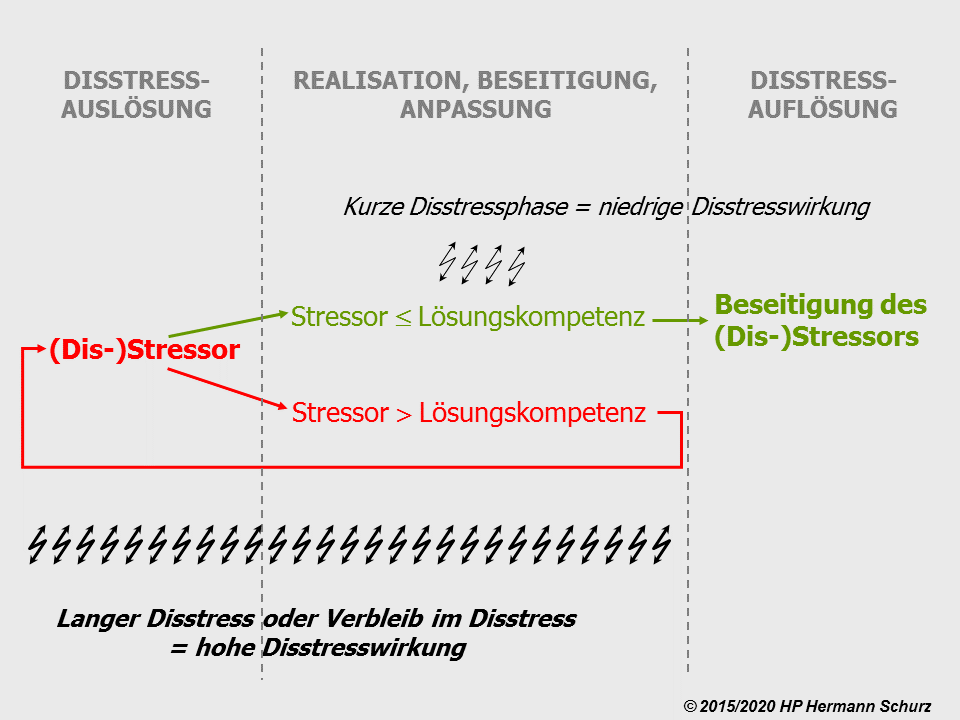
Abbildung 36: Ist die Disstresslösungskompetenz des Individuums größer als der Disstress oder entspricht sie ihm, kann es die Situation in seinem Sinne entscheiden; die Dauer der Disstressphase ist umso kürzer, je höher die individuelle Lösungskompetenz ist. Ist das Individuum von alleine nicht zur Lösung in der Lage, bleibt es in der Disstresssituation bzw. einer Endlosschleife gefangen, es sei denn, der Disstressor verschwindet von alleine oder das Individuum nimmt die Hilfe einer lösungskompetenten Person in Anspruch.
Psychosoziale Disstressoren: Wirkungsbeeinflussung durch Individualität, Stressdauer und Stressstärke
Disstressoren verursachen auf der psychisch‑emotionalen Ebene negativen Stress, der zentralnervös unter anderem in emotional relevanten Hirnarealen verarbeitet wird und als psycho‑sozialer Disstress bezeichnet wird. Das können auch abiotisch physikalisch‑chemische oder biologisch‑medizinische Stressfaktoren sein, die eine negative emotionale Wirkung entfalten. So stellt der abiotisch‑physikalische Stressor Lärm auch eine psycho‑soziale Belastung dar, ähnliches gilt für die biologisch‑medizinischen Stressoren einer chronischen Erkrankung bzw. chronischer Schmerzen.
Tabelle 19 unterscheidet psychosoziale Disstressoren in drei Lebensbereichen. Viele der Disstressoren in Tabelle 19 sind auch für Kinder und Jugendliche relevant, jedoch kommen in frühen Lebensphasen weitere dazu. Die Stressproblematik bei Kindern und Jugendlichen wird in Abschnitt 4.12.4 erörtert.
Sowohl eine depressive Verstimmung als auch eine klinische Depression gelten als eigenständige Stressoren, deren zusätzlich krank machende psychosoziale Mechanismen in Abschnitt 4.12.2 detaillierter erörtert werden.
Die Wirkungen der in Tabelle 19 genannten ‑ von außen einwirkenden ‑ Disstressoren sind sehr individuell. Sie hängen von den persönlichen Eigenschaften der betroffenen Person ab.
TABELLE 19: BEISPIELE EXTERNER PSYCHOSOZIALER DISSTRESSOREN IM ERWACHSENENALTER
Familie + Partnerschaft |
Beruf + Ausbildung |
Sonstiges Umfeld + Allgemein |
|---|---|---|
| Innerfamiliäre Konflikte - Paarkonflikte - Familiäre Konflikte - Sexuelle Gewalt in der Ehe - Trennung - Scheidung - Verpflichtungsgefühle - Auszug erwachsener Kinder Sonstige Ereignisse - Eheschließung - Kinderzuwachs - Krankheit Familienmitglied - Tod Familienmitglied |
Arbeitstätigkeit und -organisation - Unbefriedigende Arbeitstätigkeit - Überforderung - Unterforderung - Perfektionismus - Termindruck - Erfolgsdruck - Verantwortungsbereichsveränderungen - Reorganisationsmaßnahmen - Vorgesetztenwechsel - Wechseln der Fachabteilung - Mehrarbeit - Schichtarbeit - Monotone Tätigkeit - Fließbandarbeit - Unterbezahlung - Niedriglohnarbeit - Selbstständigkeit - Ruhestand Personelle Konflikte - Konflikte mit Kollegen/Untergebenen - Konflikte mit Vorgesetzten - Verpflichtungsgefühl - Mobbing - Konkurrenzdenken Arbeitsplatzsicherheit - Arbeitsplatzwechsel - Drohender Arbeitsplatzverlust - Abmahnung - Arbeitsplatzverlust - Langzeitarbeitslosigkeit Ausbildung - Ausbildungsaufnahme - Ausbildungswechsel - Ausbildungsabbruch - Anstehende Prüfung - Negative Prüfungserfahrung - Ausbildungsabschluss |
Soziale Konflikte - Bilaterale Konflikte - Spannungen in sozialen Gruppen - Gruppenzwänge - Soziale Isolation - Mobbing - Fehlende Akzeptanz des pers. Umfelds - Ständige Entwertungen durch Andere - Verpflichtungsgefühle - Normabweichende Sexualpräferenz - Geschlechtsidentitätsprobleme - Religionsbasierende Konflikte Gesundheitliche Belastungen - Schlafstörungen - Chronische Schmerzen - Chronische Erkrankungen - Lebensbedrohende Erkrankungen - Depressive Verstimmung - Depression - Suchtproblematiken Finanzielle Belastungen - Armut - Geldprobleme - Überschuldung - Aufnahme eines größeren Kredits Bedeutende Negativereignisse - Freiheitsentzug/Haft - Rechtsstreit/Gerichtsverfahren - Tod einer nahestehenden Person - Sexueller Missbrauch/Vergewaltigung - Folter - Stalking - Bedrohungen durch Dritte - Obdachlosigkeit - Naturkatastrophen - Unfälle - Kriegshandlungen Sonstiges / Innere Konflikte - Niedriges Selbstwertgefühl - Selbstentwertung - Normabweichende Sexualpräferenz - Geschlechtsidentitätsprobleme - Religionsbedingte innere Konflikte - Zeitdruck - Tageslärm - Nachtlärm - Reizüberflutung - Urbane Hektik - Umzug: Wohnungswechsel - Umzug: Wohnortwechsel |
Tabelle 19: Eine beispielhafte Auswahl psychosozialer Stressoren ohne Allgemeingültigkeit. Was für den einen Stress bedeutet, kann für den anderen stressneutral sein. Hier sind u. a. der individuelle Charakter und die individuelle Resilienz entscheidend.
Individuelle Eigenschaften beeinflussen die Wirkungen externer Disstressfaktoren. Neben der schon erwähnten Lösungskomptenz spielt auch die Resilienz eine große Rolle. Beides entwickelt und entfaltet sich jedoch nicht in einem „luftleeren Raum“, sondern ist ‑ wie jede Gehirnleistung ‑ substanzgebunden (→ Abschnitt 1.5 zur Resilienz). Somit sind Funktionalitäten und Strukturen von Hirnarealen, die über individuelle Eigenschaften bestimmen, immer mitentscheidend an der Wirkungsstärke eines Disstressors.
Bei der Disstresswirkung spielen zeitliche Aspekte eine weitere Rolle, und mit ansteigender Konfliktdauer verstärken sich die negativen Stresswirkungen (→ Abbildung 36). Es ist zu unterscheiden zwischen...
- einmalig-akutem Disstress,
- episodischem bzw. periodischem Disstress und
- chronischem Disstress.
Stress wird als chronisch betrachtet, wenn eine Änderung des Zustandes bzw. die Beseitigung des Disstressors nicht absehbar ist bzw. eine solche Änderung nur durch ein aktives Eingreifen möglich wäre, dieses aber nicht erfolgt.
Die Disstressintensität ist ebenfalls von Bedeutung, und es leuchtet unmittelbar ein, dass ein solches Merkmal existiert - auch wenn eine objektive Intensitätsmessung kaum möglich ist. Das bedeutet: Auch kurzfristiger Stress kann ‑ bei entsprechender Intensität ‑ schädigend sein. Die Intensität negativen Stresses kann auch durch mehrere parallel einwirkende Disstressoren erhöht werden.
4.12.2 Wirkungen von externem Disstress auf Gehirn und Körper
Da Disstress im Gehirn entsteht, das sämtliche psychischen und körperlichen Reaktionen als oberste Instanz steuert, betreffen Disstresswirkungen potentiell den gesamten Körper.
Über die Reizweiterleitung der Sinnesorgane erreichen die externen Informationen das Gehirn und setzen unwillentlich eine Kaskade physiologischer Aktionen in Gang, um Körper und Psyche schnell auf die besondere Situation einzustimmen. Im Zentralnervensystem treten sensorische, affektrelevante und kognitive Hirnareale in Aktion, beispielsweise Hypothalamus, Hippocampus, Amygdala oder Bereiche des Gyrus cinguli.
Stressachsen und Stresssysteme im Überblick
Zwei Stressachsen und ein ergänzendes ZNS-internes Stresssystem parieren Disstress auf unterschiedliche Weise.
Stressachse Nr. 1 wird mit zwei Teilsystemen A und B indirekt über den Blutkreislauf und Hormone gesteuert. Stressachse Nr. 2 besteht aus einem System, das direkt über Nervenreize reguliert wird.
Beide Stressachsen mit ihren insgesamt drei Systemen werden durch ein viertes ZNS-internes Stresssystem ergänzt.
Es ist derzeit unklar, ob darüber hinaus weitere Systeme existieren.
Immer spielt der Hypothalamus seine Rolle als oberstes Regelungsorgan und „Stresswächter“. Im Hypothalamus befindet sich der Nucleus paraventricularis, vermutlich die Hauptschaltstelle der zentralnervösen Stressverarbeitung. Hypothalamus bzw. Nucleus paraventricularis erhalten ihre Informationen aus verschiedenen Großhirnarealen, vor allem von Hippocampus, Amygdala und Gyrus cinguli.
Die Hormonausschüttung in den Blutkreislauf wird bei beiden Systemen der Stressachse Nr.1 von Hypothalamus und Hypophyse gemeinsam geregelt. Die Systeme unterschieden sich durch ihre Zielorgane. Zielorgan des ersten Systems ist die Nebennierenrinde, Zielorgan des zweiten die Schilddrüse:
- Stressachse 1 A bzw. System 1: Hypothalamus/Hypophyse‑Nebennierenrinden (Hyt/Hyp‑NNRinde)
- Stressachse 1 B bzw. System 2: Hypothalamus/Hypophyse‑Schilddrüsen (Hyt/Hyp‑SD)
Das dritte System steuert der Hypothalamus direkt via sympathischem und parasympathischem Nervensystem (Teile des vegetativen Nervensystems VNS) und nutzt das Nebennierenmark zur Hormonfreisetzung unter Umgehung des Blutkreislaufs. Dabei erfolgt die Aktivierung über den Sympathikus, während der Parasympathikus für drosselnde Aktivitäten genutzt wird:
- Stressachse 2 bzw. System 3: Hypothalamus‑VNS‑Nebennierenmark (Hyt‑S/P‑NNMark)
Ein viertes System sorgt für weitere Anpassungen, die mit den drei vorher genannten Mechanismen nicht zu bewältigen sind:
- System 4: ZNS‑internes Hypothalamus/Hypophysen‑Stresssystem (ZiS‑Hyt/Hyp)
Stresssystem 1: Hypothalamus/Hypophyse-Nebennierenrinden (Hyt/Hyp-NNR)
Der Mechanismus steuert vor allem die Glucocorticoidsynthese und -abgabe in die Blutbahn, vorwiegend das als Stresshormon bezeichnete Körperhormon Cortisol.
Cortisol ist von der Blutzuckerregulation bekannt (→ Abschnitt 4.8), es soll Glukosereserven mobilisieren und die Gluconeogenese in Leber und Nieren erhöhen. In Stresssituationen ist ein hohes Energieangebot durch Glukose überlebensnotwendig.
Cortisol hat darüber hinaus für eine stressangepasste Psyche zu sorgen und darf daher die Blut-Hirn-Schranke zum Gehirn durchbrechen. Das Hormon wirkt dort ähnlich wie ein Neurotransmitter: Cortisol erhöht die geistige Leistungsfähigkeit, indem es Gestresste in die Lage versetzt, Aufmerksamkeit zu bündeln und sich völlig auf die schwierige Situation zu konzentrieren.
Auch auf das Immunsystem hat Cortisol einen Einfluss. Bei kurzfristigem Stress, der nicht länger als wenige Stunden anhält, stärkt es die Immunabwehr. Leider verkehrt sich dieser Effekt bei länger anhaltendem Stress in das Gegenteil.
Stresssystem 1 ist durch die Hormonkaskade CRH/ADH → ACTH → Cortisol charakterisiert. Bei Stress sezerniert der Hypothalamus das Freisetzungshormon Corticotropin Releasing Hormon CRH und das antidiuretische Körperhormon ADH.
CRH wird direkt lokal vom Hypothalamus an die Hypophyse via Nervenbahnen weitergegeben und regt ‑ mit Unterstützung von ADH ‑ die Hypophyse zur Produktion und Ausschüttung des adrenocorticotropen Hormons ACTH ins Blut an. ACTH erreicht via Blutbahn die Nebennierenrinden, die daraufhin innerhalb weniger Minuten Glucocorticoide bzw. Cortisol ausschütten. Damit reagiert dieses Stresssystem insgesamt eher langsam und träge in Anbetracht der Tatsache, dass Stressreaktionen in Sekunden zu erfolgen haben, um ihren Zweck zu erfüllen.
Das durch den Hypothalamus ins Blut sezernierte antidiuretische Körperhormon ADH senkt darüber hinaus das Urinvolumen, indem es die Nieren anregt, vermehrt Wasser aus dem Primärharn rückzugewinnen. Dadurch wird der lästige Harndrang minimiert, was einer erfolgreichen Stressbewältigung entgegenkommt.
Konsequenzen für die Regelkreissteuerung ergeben sich durch den Umstand, dass Cortisol die Blut-Hirn-Schranke mühelos passiert. Dadurch erreicht Cortisol automatisch auch den Hypothalamus, der das System mittels negativer Rückkopplung steuert: Erhöhen sich die Cortisolwerte im Blut, wird dies von dessen Glucocorticoidrezeptoren registriert, der daraufhin bei einer zum Parieren des Stresses ausreichenden Cortisolmenge die Produktion von CRH und ADH drosselt, was wiederum über die Senkung des Hypophysenhormons ACTH die Nebennieren veranlasst, weniger Cortisol auszuschütten.
Kurzfristig kann Cortisol in gewissen Grenzen CRH und ACTH direkt senken und damit seine eigene Produktion sehr schnell bei Bedarf drosseln („Ultra-Short-Feedback“).
Einen weiteren hemmenden Einfluss auf die Cortisolproduktion hat das vorwiegend von den Herzmuskelzellen (und in kleineren Mengen auch in den Nebennieren) produzierte atriale natriuretische Peptid ANP, denn es unterdrückt die ADH-Ausschüttung im Hypothalamus. Weniger ADH reduziert darüber hinaus die stressbedingt gesteigerte Nierenfiltrationsrate, so dass wieder mehr Urin gebildet wird.
Neben der Hauptaufgabe als Cortisolsyntheseregulierer könnten mit Stresssystem 1 weitere Effekte bei Stress mit den Hormonen bzw. Peptiden CRH, ADH und ANP verbunden sein.
Das Freisetzungshormon CRH könnte eine eigenständige Bedeutung als zentralnervöser Botenstoff haben, denn es wurden in zahlreichen affektrelevanten Hirnarealen CRH-Rezeptoren gefunden. So führt Andreas Ströhle vom Max‑Planck‑Institut für Psychiatrie in München aus: „Die Effekte von CRH werden durch zwei spezifische, G-Protein-gekoppelte Rezeptoren, den CRH‑R1 und den CRH‑R2, vermittelt. Der CRH‑R1 wird nahezu ausschließlich im frontalen Kortex, dem basalen cholinergen Vorderhirn, den cholinergen Kernen des Hirnstamms, dem Collicolus superior, der basolateralen Amygdala, dem Zerebellum, dem N. trigeminus und dem Hypophysenvorderlappen gefunden. Dagegen ist der CRH‑R2 stärker im PVN, dem lateralen Septum, den zentralen und medialen Teilen der Amygdala sowie dem serotoninergen Nucleus Raphe vorhanden. Eine gemischte Rezeptorexpression findet sich im Bulbus olfactorius, dem Hippokampus, dem entohirnalen Kortex, der Stria terminalis und dem periaqueduktalen Höhlengrau. Bemerkenswerterweise findet sich nur eine gering bis mäßig ausgeprägte Expression von CRH‑R1 in der zentralen Amygdala und der Substantia nigra. Ebenfalls könnte CRH im Hypothalamus selbst wirksam sein. Es gibt Forschungen, die einen direkten Zusammenhang zwischen einer Depression und CRH belegen.“ (Quelle: A. Ströhle, Die Neuroendokrinologie von Stress und die Pathophysiologie und Therapie von Depression und Angst, 2003, Der Nervenarzt, 3/2003, S. 279 ‑ 292, Springer‑Verlag, Berlin).
In einer ähnlichen Weise könnte das im Nucleus paraventricularis des Hypothalamus sezernierte ADH eine direkte Rolle in der Stressregulation spielen, da es sich in der Umgebung innerhalb der affektiven Areale verteilt. Es könnte damit sowohl im Hypothalamus selber als auch in dessen Umgebung für physiologische Veränderungen ‑ vergleichbar mit denen durch CRH ‑ sorgen. Hier gibt es leider noch sehr wenig wissenschaftlich verlässliche Erkenntnisse.
ANP wurde schon beim Rückkopplungsmechanismus der Cortisolproduktion erwähnt. Dafür ist aber das peripher in den Herzmuskelzellen und Nebennieren produzierte ANP zuständig, wobei es, die Blut‑Hirn‑Schranke überwindend, seine regulatorischen Aufgaben zentralnervös auf allen Ebenen in Hypothalamus, Hypophyse und weiteren affektrelevanten Hirnregionen ausführt. ANP wird aber auch in kleineren Mengen im Gehirn ‑ hauptsächlich im Hypothalamus ‑ synthetisiert. Man fand Rezeptoren u. a. im periventrikulären und paraventrikulären Hypothalamus, der zentralen Amygdala und in einigen der Formatio reticularis zugehörigen Nervenbahnen im Hirnstamm (Quelle: A. Ströhle, 2003, wie oben).
Im Hypothalamus ist ANP auch für die Durstreduktion zuständig. Ob diese Aktivitäten durch das peripher oder zentral synthetisierte ANP ausgelöst werden, ist derzeit nicht bekannt. Andreas Ströhle führt in seinem schon oben erwähnten Beitrag aus, dass für ANP eine dämpfende, angstlösende Eigenschaft nachgewiesen wurde: „Neben der Hemmung des Stresshormonsystems konnten wir auch eine anxiolytische Aktivität von ANP im Tierexperiment (...) wie auch beim Menschen beschreiben.“ (Quelle: A. Ströhle, 2003, wie vorher). Es spricht einiges dafür, dass sich zentralnervöses als auch peripheres ANP in ihrer Wirkung eher ergänzen, denn unterschiedliche Rollen bei durchlässiger Blut-Hirn-Schranke sind schwer vorstellbar.
Die wissenschaftlichen Kenntnisse über Stresssystem 1 sind immer noch relativ gering und die obige Beschreibung des Mechanismus ist stark vereinfacht. Wer an einer detaillierteren Darstellung interessiert ist, kann sich auf der Webseite des Projekts ADxS.org ausführlich informieren (Quelle: Die HPA‑Achse/Stressregulationsachse, ADxS.org ‑ Das AD(H)S-Kompendium, https://www.adxs.org/stress-und-adhs/...). Das nichtkommerzielle Projekt ADxS.org setzt sich intensiv mit der ADHS‑Erkrankung auseinander, denn hier spielt Stress eine wichtige Rolle.
Stresssystem 2: Hypothalamus/Hypophyse-Schilddrüse (Hyt/Hyp-SD)
Zielorgan und Zielwirkstoff des zweiten hormongesteuerten Stresssystems sind die Schilddrüsen und die von ihnen gebildeten Körperhormone Triiodthyronin (T3) und Thyroxin (T4).
Wie Cortisol sorgen auch T3 und T4 für Wachheit, Konzentrationsfähigkeit und gesteigerten Antrieb. Um die körperlichen Voraussetzungen der Stressbewältigung zu schaffen, erhöhen sie zusätzlich den Stoffwechselgrundumsatz.
Die T3/T4‑Synthese wird ebenfalls via Hypothalamus gesteuert, diesmal mittels des Freisetzungshormons Thyrotropin Releasing Hormon TRH. TRH regt die Hypophyse zur Synthese des Thyreoidea‑stimulierenden Hormons TSH an, das via Blutbahn die Schilddrüse erreicht und sie veranlasst, Triiodthyronin und Thyroxin zu bilden:
TRH → TSH → Triiodthyronin/Thyroxin (T3/T4)
Da T3 und T4 im Gehirn ebenfalls wichtige Funktionen erfüllen, stellt die Blut‑Gehirn‑Schranke auch hier kein Hindernis dar, und der Mechanismus steuert sich durch negative Rückkopplung via Hypothalamus und drosselt die T3/T4‑Produktion situationsgerecht.
Auch hier gibt es weitere Rückkopplungsschleifen, beispielsweise kann TSH auf die eigene Synthese Einfluss nehmen. Sinn der zusätzlichen Steuerungsmöglichkeit ist auch hier der Bedarf nach kurzfristigen Steuerungsmechanismen („Ultra‑Short‑Feedback“).
Außerdem stimulieren die Schilddrüsenhormone den Sympathikus und unterstützen damit den Hypothalamus bei der Steuerung der zweiten Stressachse (→ Stresssystem 3 im folgenden Abschnitt).
Auch Stresssystem 2 reagiert durch die Nutzung des Blutkreislaufs zur Regelsteuerung relativ langsam.
Stresssystem 3: Hypothalamus-Vegetatives Nervensystem-Nebennierenmark (Ht-VNS-NNM)
Bei Stress stimuliert der Hypothalamus mittels Sympathikus bzw. Parasympathikus des vegetativen Nervensystems direkt die Produktion der Körperhormone Adrenalin und Noradrenalin im Nebennierenmark. Im Gegensatz zu den Stresssystemen 1 und 2 wird hier also kein Releasing-Hormon sezerniert, und die Hypophyse gibt daher auch kein Hormon in den Blutkreislauf, um das Zielorgan zur Körperhormonausschüttung anzutreiben. Im Gegensatz zu diesen Systemen reagiert Stresssystem 3 sehr schnell innerhalb von Sekunden.
Die wichtigste hormonelle Wirksubstanz des dritten Stresssystems ist Adrenalin, während Noradrenalin lediglich eine unterstützende Funktion hat. Beide Substanzen haben die Aufgabe, knappe Körperressourcen auf die zur Stressbewältigung bedeutenden Organe zu verlagern, hauptsächlich durch eine Veränderung von Mikrodurchblutung, glatter Muskulatur und Energieversorgung. Die betroffenen Organe bzw. Organsysteme sind Herz/Kreislauf, gestreifte Muskulatur, Verdauung, Harn- und Geschlechtsapparat, Atmung, Energieversorgung und die Augen:
- Arteriolenverengung mit Wirkungen auf Muskulatur, Herz-Kreislauf und das urogenitale System
Adrenalin und Noradrenalin sorgen für die Verengung kleinerer Blutgefäße (Arteriolen) in den für Stress unwichtigen bzw. kontraproduktiven Körperregionen, zum Beispiel in Haut, Geschlechtsorganen oder Nieren, deren Durchblutung daraufhin sinkt.
Durch die Minderversorgung des größten Organs, der Haut, bleibt viel Blutvolumen für wichtigere Aufgaben übrig. Der/die Gestresste wird im Akutzustand dann blass und „Weiß wie die Wand“. Parallel dazu steigert Adrenalin die Mikrodurchblutung stresswichtiger Regionen, vor allem der gestreiften Skelettmuskulatur, die zur Durchführung körperlicher Stressreaktionen benötigt wird.
Adrenalin erhöht darüber hinaus Herzfrequenz und Herzkontraktionsfähigkeit, auch mittels Erweiterung der Herzkranzgefäße. Eine besonders unangenehme Nebenwirkung von Herzleistungsmobilisierung und Blutgefäßverengung in stressunwichtigen Körperregionen ist die Erhöhung des Blutdrucks.
Eine Minderdurchblutung der Nieren führt zu einer generellen Senkung des Urinvolumens - und damit zu einem Rückgang des Harndrangs; dadurch unterstützt Stresssystem 3 das erste System, durch dessen Filtrationsratenerhöhung das Urinvolumen ebenfalls absinkt.
- Glatte Muskulatur mit Auswirkungen auf Atmung, Verdauung und das urogenitale System
Durch die adrenalinbedingte Erschlaffung oder Kontraktion der glatten Muskulatur werden die Leistungen nicht stressrelevanter Organe reduziert bzw. die Leistungen stressrelevanter Organe erhöht.
Die glatte Bronchienmuskulatur erschlafft und führt so zu einer Optimierung der Atmung mit einhergehender Verbesserung der Lungenventilation, Zunahme des Atemvolumens und Erhöhung der Atemfrequenz. Wahrscheinlich wird auch die für die Atmung zuständige Stammhirnregion aktiviert, was indirekt über eine Verbesserung der Hirndurchblutung bzw. direkt durch den Hypothalamus oder beides veranlasst wird.
Im Verdauungstrakt führt die Muskelerschlaffung zu einer Verlangsamung von Magen- und Darmbewegungen und damit der Verdauungsprozesse. Ergänzt wird das durch den Rückgang des Speichelflusses (viele Menschen kennen das aus eigener Erfahrung, wenn sich schon ab der ersten Schrecksekunde der Mund staubtrocken anfühlt).
Die adrenalinbedingte Kontraktion des glatten Blasenschließmuskels senkt auf eine weitere Weise den Harndrang und unterstützt damit schon erwähnte vergleichbare Reaktionen.
- Energieversorgung und Glukoseaufnahme in der Muskulatur
Adrenalin erhöht den allgemeinen Energieumsatz, den Abbau von Fett- und Glukosereserven und die Neubildung von Glukose. Passend dazu erhöht sich die Glukoseaufnahme der gestreiften Muskelzellen (→ Abschnitt 4.8).
Adrenalin ergänzt dadurch die Cortisol- bzw. Triiodthyronin- und Thyroxinaktivitäten der Stresssysteme 1 und 2, die ebenfalls Energiereserven mobilisieren.
- Visuelles System
Eine Pupillenerweiterung um etwa 10% soll die Sehfähigkeit bei Stress erhöhen, vor allem in der Nacht.
Adrenalin und Noradrenalin der Nebennieren aktivieren ausschließlich verschiedene peripheren Körperorgane und Prozesse, im Gehirn haben die Nebennierenhormone keinerlei Funktionen und dürfen auch nicht dorthin gelangen. Das Gehirn produziert selber Noradrenalin und ‑ in geringen Mengen ‑ Adrenalin, die allerdings nicht hormonell wirken, sondern als Neurotransmitter agieren. Insbesondere stellt das im Gehirn produzierte Noradrenalin einen wichtigen neuronalen Botenstoff dar. Daher wäre es fatal, wenn die beiden Nebennierenhormone ins Gehirn gelängen, dort unerwünschte Wirkungen entfalteten und die dortigen Botenstoffsyntheseprozesse durcheinanderbrächten.
Adrenalin und Noradrenalin des Nebennierenmarks wirken zentralnervös nur indirekt reflektorisch durch ihre peripheren Körperfunktionen, zum Beispiel durch die Erweiterung der Hirngefäße oder den Anstieg des Blutzuckers.
Um die Abschottung des Gehirns von den Nebennierenmarkhormonen gewährleisten zu können, muss die Blut‑Hirn‑Schranke allerdings optimal funktionieren. Leider ist das nicht immer der Fall. Eine durchlässige Blut-Hirn-Schranke hat einen eigenständigen Krankheitswert (→ weiter unten).
Da beide Substanzen unter normalen Umständen nicht ins Gehirn gelangen, können sie auch den Hypothalamus nicht erreichen, so dass es im Stresssystem 3 keine negative Rückkopplung gibt, was ein weiterer Unterschied zu den ersten beiden Stresssystemen ist. Auch die Regelung des dritten Stresssystems erledigt der Hypothalamus daher ausschließlich direkt via vegetativem Nervensystem (VNS) mittels Sympathikus bzw. Parasympathikus.
Stresssystem 4: ZNS-internes Hypothalamus-Hypophysen-Stresssystem (ZiS-Hyt/Hyp)
Aufgaben des vierten Stresssystems ist die Ausschüttung von (Beta‑)Endorphin, mit dem Schmerzempfinden, Hunger, Durst und Sexualtrieb gesenkt, und die Stresstoleranz über den euphorischen Effekt erhöht wird. Durch eine Steigerung von Angst soll das Individuum zu selbstschützenden Aktivitäten angeregt werden. Endorphine werden in Hypothalamus und Hypophyse gebildet; es ist nicht klar, ob Hypophyseaktivitäten völlig unabhängig von Aktivitäten des Hypothalamus geschehen.
Bei der Diskussion um die Folgen negativen Stresses werden die Mechanismen dieses Systems trotz ihres großen Einflusses auf Gestresste meist wenig beachtet.
Auswirkungen von Disstress auf zentralnervöse und periphere Strukturen in der graphischen Übersicht
Abbildung 37 stellt die vier Stresssysteme in einer Graphik dar.
Auch wenn die Abbildung dadurch zunächst unübersichtlich wirkt, macht sie dennoch die mannigfaltigen physiologischen Interaktionen und Zusammenhänge auf einen Blick deutlich.
ABBILDUNG 37: UNMITTELBARE PHYSIOLOGISCHE REAKTIONEN AUF DISSTRESS
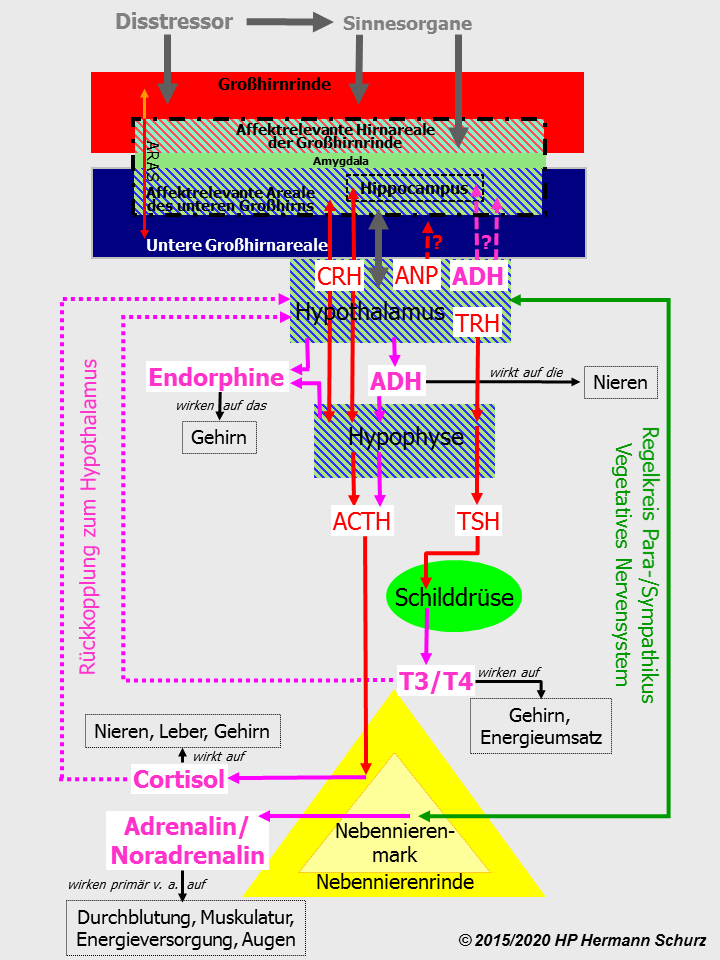
Abbildung 37: Stresssignale erreichen das Gehirn primär über Sinnesorgane. Vier Stresssysteme sorgen für die Bereitschaft von Körper und Psyche, Stress zu beseitigen bzw. zu parieren. System 1 sorgt für die Ausschüttung des Nebennierenrindenhormons Cortisol, System 2 für die Ausschüttung der Schilddrüsenhormone T3 und T4. Ausschüttung und Steuerung erfolgen in beiden Systemen auf eine ähnliche Weise durch Releasing Hormone und Hypophysenhormone, sie werden daher derselben Stressachse zugerechnet. Demgegenüber wird die Ausschüttung von Adrenalin und Noradrenalin im Nebennierenmark über das vegetative Nervensystem gesteuert. In einem vierten Stresssystem schütten Hypothalamus und Hypophyse Endorphine aus, die im Gehirn weitere wichtige Funktionen bei der Stressbewältigung erfüllen.
Zur Problematik erhöhter aggressiv-reaktiver Sauerstoff-Spezies (ROS) bei psychosozialem Stress
Wo Hormone produziert werden und wirken, müssen sie auch inaktiviert und abgebaut werden. Das geschieht im Nervensystem durch enzymatisch-lysosomalen Abbau. Hormone, die sich schon im Inneren der Zelle befinden, weil sie an zellinneren Rezeptoren binden, werden dort abgebaut. Hormone, die an Rezeptoren der äußeren Zellmembran binden, werden mit Hilfe des Endozytose-Prozesses ins Zellinnere geschleust und dort durch Lysosomen beseitigt (Autophagie).
Werden in Stresssituationen viele Hormone produziert, müssen auch entsprechend mehr Hormone inaktiviert und abgebaut werden. Dadurch kommt es zu erhöhtem oxidativen Zellstress, denn beim Hormonabbau entstehen zwei Arten aggressiver reaktiver Sauerstoff‑Spezies (ROS): freie Sauerstoffradikale und aggressive Sauerstoffverbindungen. ROS können allen Zellgeweben ‑ insbesondere der DNA und inneren bzw. äußeren Membranen ‑ gefährlich werden.
Eine weitere Gefahr durch aggressive ROS resultiert aus dem stressbedingt erhöhten Energiebedarf des Gehirns. Denn die vermehrte Bereitstellung von Energie führt zu einem generellen Anstieg des Sauerstoffverbrauchs in der zellulären Atmungskette und damit auch zu einem entsprechend hohen Ausstoß schädlicher ROS im Gehirn.
Viel psychosozialer Stress bedeutet demnach auch eine übermäßige Produktion aggressiver Substanzen, sowohl im Zentralnervensystem als auch in peripheren Organen. Mehr über ROS und deren zellschädigende Mechanismen werden im Abschnitt 4.13 erörtert. Die dort gezogenen Schlussfolgerungen hier aber schon mal vorweg: Psychosozialer Stress führt zu zusätzlichen aggressiv-oxidativen Zellprozessen im Zentralnervensystem. Bei Stress spielen sich diese belastenden Vorgänge hauptsächlich in emotionsrelevanten Hirnarealen ab, die auch an der Entstehung affektiver Erkrankungen beteiligt sind.
Damit lässt sich eine direkte Ursachen-Wirkungs-Kette zwischen Stress und stressbedingten krankhaften Gehirnveränderungen ableiten: Stress schädigt im Gehirn Nervenzellen auch durch eine erhöhte Anzahl reaktiver Sauerstoff-Spezies (ROS).
Störungen der Blut-Hirn-Barriere durch psychosozialen Stress
Eine weitere Folge psychosozialen Stresses könnte in einer möglichen Erhöhung der Durchlässigkeit bzw. Permeabilität der Blut-Hirn-Schranke (BHS) bestehen. In diesem Falle würden vor allem bei Stress unerwünschte und schädliche Substanzen vermehrt das Gehirn erreichen.
So führt Franz Schneider in seinem Buch über cerebrale Leistungsförderung aus: „Zu den Faktoren, welche die normale Permeabilität der Blut-Hirn-Schranke gefährden, zählen nach Bested et al. (2001), Kuschinsky (2001) und Thiel et al. (2001) u. a. (...) Streß (...). Es ist möglich, daß ein Zusammenbruch der Permeabilität der BHS zu zellulären Dysfunktionen und Störungen der neuronalen Übertragung im ZNS führt. (...) Das Konzept der BHS hat sich im Verlauf der letzten Jahre von einer passiven und relativ unveränderlichen Gewebestruktur zu einer dynamischeren Schnittstelle zwischen Blut und Gehirngewebe weiterentwickelt (Lataste, 1992).“ (Quelle: Franz J. Schneider, Gehirn, Gesundheit, Gymnásion, Cuvilier Verlag Göttingen, Göttingen 2008).
Die Veränderungen der Blut-Hirn-Schranke könnten durch übermäßig vorhandene Stresshormone oder andere stressbedingte Substanzen verursacht werden. Ebenfalls ist der erhöhte Bedarf des gestressten Gehirns an Mikro- und Makronährstoffen und anderen Substanzen, beispielsweise Glukose oder Aminosäuren, ein potentieller Grund dafür, dass die Schleusen der Blut-Hirn-Schranke weiter geöffnet werden und damit ‑ neben erwünschten Substanzen ‑ leider auch viele unerwünschte Stoffe das Gehirn erreichen.
Die genauen Ursachen stressbedingter Veränderungen an der Durchlässigkeit der Blut-Hirn-Schranke sind weitgehend noch Gegenstände der Forschung.
4.12.3 Psychosozialer Stress als externer Auslöser Affektiver Störungen
Ein Kausalzusammenhang zwischen Stress als verursachendem externen Faktor und affektiven Erkrankungen gilt aufgrund zahlreicher, darauf hindeutender Forschungsergebnisse als belegt. Im Abschnitt 4.12.5 weiter unten werden diese zusammen mit Studien noch vorgestellt.
Trotz vieler Erkenntnisse, beispielsweise über verschiedene Stressregulationssysteme, Rezeptoren oder Substanzen, konnten die zugrundeliegenden Mechanismen noch nicht völlig geklärt werden. Ein allgemein akzeptiertes und umfassendes Stressmodell existiert nicht.
Sicher ist: Bei Stress wird das Gehirn multifaktoriell durch verschiedene Prozesse und körpereigene Substanzen belastet. Es ergibt daher auch keinen Sinn, die Ursache für die Pathologisierung der Stresssysteme und für stressbedingte Affekterkrankungen in einzelnen Prozessen oder Substanzen zu suchen.
Auswirkungen von Disstress auf stressrelevante Organe bzw. Organsysteme
Unter Zuhilfenahme des kausaltheoretischen Modells sind Zusammenhänge zwischen Stress und Affekterkrankungen plausibel nachvollziehbar. Stress ist demnach ein (weiterer) negativer externer Faktor, der zu ungünstigen Veränderungen in affektrelevanten Hirnarealen führen kann (→ Abschnitt 1.5).
Tabelle 20 zeigt alle bei der Stressabwehr beteiligten ZNS-Areale, sonstige Akteure, Substanzen und potentielle pathologische Veränderungen. Ungeachtet der mit dieser tabellarischen Darstellung verbundenen Redundanzen sind alle Möglichkeiten aufgeführt, auch um Komplexität und Umfang der Auswirkungen psycho‑sozialen Disstresses aufzuzeigen.
Es ist zu beachten, dass sowohl Neuronen und teilweise auch Gliazellen betroffen sein können. Darüber hinaus könnten auch die wichtigen Gefäße zur Versorgung des Gehirns mit Sauerstoff und Nährstoffen durch Stress auf vergleichbare Weise geschädigt werden mit entsprechenden negativen Folgen für die Hirnareale.
TABELLE 20: AKTEURE, AKTIVITÄTEN UND FOLGEN DER STRESSVERARBEITUNG
Organ(-system) |
Potentielle Aktivitäten/ Änderungen bei Stress |
Potentielle Schädigungen durch Dauerstress im Detail |
|---|---|---|
| Amygdala | Aktivitätsänderungen - Zunahme der Aktivitäten - Abnahme der Aktivitäten Strukturelle Änderungen - Volumen - Neuronenvernetzung - Rezeptoren |
Langandauernde Aktivitätsveränderungen durch ADH Langandauernde Aktivitätsveränderungen durch ANP Langandauernde Aktivitätsveränderungen durch Cortisol Langandauernde Aktivitätsveränderungen durch CRH Volumenänderungen (z. B. Hypotrophie, Hypertrophie) Dauerhafte Veränderungen der neuronalen Vernetzung Dauerhafte Veränderungen an ADH-Rezeptoren Dauerhafte Veränderungen an ANP-Rezeptoren Dauerhafte Veränderungen an Glucocorticoid-Rezeptoren Dauerhafte Veränderungen an CRH-Rezeptoren Hormonabbau produziert Hirnzellen schädigende ROS Hoher Energieumsatz bedingt mehr Hirnzellen schädigende ROS |
| Formatio reticularis | Aktivitätsänderungen - Zunahme der Aktivitäten - Abnahme der Aktivitäten Strukturelle Änderungen - Volumen - Neuronenvernetzung - Rezeptoren |
Langandauernde Aktivitätsveränderungen durch ADH Langandauernde Aktivitätsveränderungen durch ANP Langandauernde Aktivitätsveränderungen durch Cortisol Langandauernde Aktivitätsveränderungen durch CRH Volumenänderungen an Kernen der Formatio reticularis Dauerhafte Veränderungen der retikulären Vernetzung Dauerhafte Veränderungen an ADH-Rezeptoren Dauerhafte Veränderungen an ANP-Rezeptoren Dauerhafte Veränderungen an Glucocorticoid-Rezeptoren Dauerhafte Veränderungen an CRH-Rezeptoren Hormonabbau produziert Hirnzellen schädigende ROS Hoher Energieumsatz bedingt mehr Hirnzellen schädigende ROS |
| Hippocampus | Aktivitätsänderungen - Zunahme der Aktivitäten - Abnahme der Aktivitäten Strukturelle Änderungen - Volumen - Neuronenvernetzung - Rezeptoren |
Langandauernde Aktivitätsveränderungen durch ADH Langandauernde Aktivitätsveränderungen durch ANP Langandauernde Aktivitätsveränderungen durch Cortisol Langandauernde Aktivitätsveränderungen durch CRH Schädigung der Nervenzellenfortsätze im Hippocampus Hippocampus-Atrophie Dauerhafte Veränderungen der neuronalen Vernetzung Dauerhafte Veränderungen an ADH-Rezeptoren Dauerhafte Veränderungen an ANP-Rezeptoren Dauerhafte Veränderungen an Glucocorticoid-Rezeptoren Dauerhafte Veränderungen an CRH-Rezeptoren Hormonabbau produziert Hirnzellen schädigende ROS Hoher Energieumsatz bedingt mehr Hirnzellen schädigende ROS |
| Hypophyse | Aktivitätsänderungen - Zunahme der Aktivitäten - Abnahme der Aktivitäten Strukturelle Änderungen - Volumen - Neuronenvernetzung - Rezeptoren |
Langandauernde Aktivitätsveränderungen durch ADH Langandauernde Aktivitätsveränderungen durch ANP Langandauernde Aktivitätsveränderungen durch Cortisol Langandauernde Aktivitätsveränderungen durch CRH Chronisch hohe ACTH-Synthese Chronisch hohe TSH-Synthese Dauerhafte Erschöpfung der ACTH-Synthese Dauerhafte Erschöpfung der TSH-Synthese Dauerhafte Veränderungen der neuronalen Vernetzung Dauerhafte Veränderungen an ADH-Rezeptoren Dauerhafte Veränderungen an ANP-Rezeptoren Dauerhafte Veränderungen an Glucocorticoid-Rezeptoren Dauerhafte Veränderungen an CRH-Rezeptoren Hormonabbau produziert Hirnzellen schädigende ROS Hoher Energieumsatz bedingt mehr Hirnzellen schädigende ROS |
| Hypothalamus | Aktivitätsänderungen - Zunahme der Aktivitäten - Abnahme der Aktivitäten Strukturelle Änderungen - Volumen - Neuronenvernetzung - Rezeptoren |
Langandauernde Aktivitätsveränderungen durch ADH Langandauernde Aktivitätsveränderungen durch ANP Langandauernde Aktivitätsveränderungen durch Cortisol Langandauernde Aktivitätsveränderungen durch CRH Chronisch hohe ADH-Synthese Chronisch hohe ANP-Synthese Chronisch hohe CRH-Synthese Chronisch hohe TRH-Synthese Dauerhafte Erschöpfung der ADH-Synthese Dauerhafte Erschöpfung der ANP-Synthese Dauerhafte Erschöpfung der CRH-Synthese Dauerhafte Erschöpfung der TRH-Synthese Dauerhafte Veränderungen der neuronalen Vernetzung Dauerhafte Veränderungen an ADH-Rezeptoren Dauerhafte Veränderungen an ANP-Rezeptoren Dauerhafte Veränderungen an Glucocorticoid-Rezeptoren Dauerhafte Veränderungen an CRH-Rezeptoren Hormonabbau produziert Hirnzellen schädigende ROS Hoher Energieumsatz bedingt mehr Hirnzellen schädigende ROS |
| Weitere affekt- relevante Hirnareale, z. B. Thalamus, Gyrus cinguli... |
Aktivitätsänderungen - Zunahme der Aktivitäten - Abnahme der Aktivitäten Strukturelle Änderungen - Volumen - Neuronenvernetzung - Rezeptoren |
Langandauernde Aktivitätsveränderungen durch ADH Langandauernde Aktivitätsveränderungen durch ANP Langandauernde Aktivitätsveränderungen durch Cortisol Langandauernde Aktivitätsveränderungen durch CRH Schädigung der Nervenzellenfortsätze im präfrontalen Cortex Volumenabnahme verschiedener affektiver Areale Volumenzunahme verschiedener affektiver Areale Dauerhafte Veränderungen der neuronalen Vernetzung Dauerhafte Veränderungen an ADH-Rezeptoren Dauerhafte Veränderungen an ANP-Rezeptoren Dauerhafte Veränderungen an Glucocorticoid-Rezeptoren Dauerhafte Veränderungen an CRH-Rezeptoren Hormonabbau produziert Hirnzellen schädigende ROS Hoher Energieumsatz bedingt mehr Hirnzellen schädigende ROS |
| Blut-Hirn-Schranke | Aktivitätsänderungen - Zunahme Permeabilität |
Diverse Schadsubstanzen erreichen das Gehirn Weitere Folgen ungeklärt bzw. Gegenstand der Forschung |
| Nebennierenmark | Aktivitätsänderungen - Zunahme der Aktivitäten - Abnahme der Aktivitäten Strukturelle Änderungen - Volumen - Rezeptoren |
Chronisch hohe Katecholamin-Synthese - Adrenalin - Noradrenalin Langfristige Erschöpfung der Katecholamin-Synthese - Adrenalin - Noradrenalin Vergrößerung des Nebennierenmarks |
| Nebennierenrinde | Aktivitätsänderungen - Zunahme der Aktivitäten - Abnahme der Aktivitäten Strukturelle Änderungen - Volumen - Rezeptoren |
Chronisch hohe Cortisol-Synthese Langfristige Erschöpfung der Cortisol-Synthese Vergrößerung der Nebennierenrinde Veränderungen der ACTH-Rezeptoren |
| Schilddrüse | Aktivitätsänderungen - Zunahme der Aktivitäten - Abnahme der Aktivitäten Strukturelle Änderungen - Volumen - Rezeptoren |
Chronisch hohe T3/T4-Synthese Langfristige Erschöpfung der T3/T4-Synthese Veränderungen der TSH-Rezeptoren |
Tabelle 20: Die Mechanismen potentieller Schädigungen durch (Dauer-)Stress im Gehirn bzw. in Körperorganen ähneln sich. Die hohe Zahl involvierter Organe bzw. Organsysteme mit zahlreichen Schädigungspotentialen verdeutlicht die Gefährlichkeit von Stress.
Tabelle 20 zeigt, dass das stressbedingte Geschehen im Gehirn und den beteiligten peripheren Organen komplex ist. Daher ist die Sicht getrübt, wenn sich Analysen der Zusammenhänge von Stress, Stressregulationsstörungen und Affekterkrankungen auf einzelne potentielle Fehlprozesse oder Substanzen beschränken.
Zur Identität stress- und affektrelevanter Hirnareale
Die Gehirnlandkarte (→ Abschnitt 1.3.1) zeigt, dass affektsteuernde Hirnareale mit stressrelevanten Arealen fast deckungsgleich sind (→ Abbildung 38 unten).
Potentiell affektrelevante Areale sind dunkel dargestellt, alle an der Stressregulation beteiligten Hirnareale mit der unterbrochenen roten Linie umrandet.
ABBILDUNG 38: AFFEKT- UND STRESSRELEVANTE HIRNAREALE IM FUNKTIONSMODELL
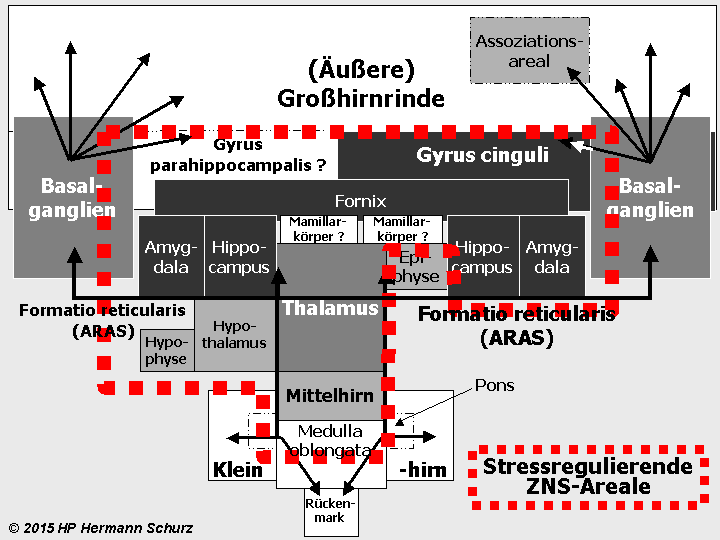
Abbildung 38: So gut wie alle in verschiedenen Grautönen markierten affektrelevanten Areale sind auch in die Steuerung und Regulierung von Stress involviert. So werden die komplexen Zusammenhänge zwischen Stress und Affektstörungen kausaltheoretisch nachvollziehbar. Stress als (mit‑)verantwortlicher Auslöser für Affektstörungen ist vor allem multikausal zu erklären.
Stress, Stressregulationsstörungen und Affektstörungen: Kausale Zusammenhänge
Die Prozesse in Tabelle 20 und Abbildung 38 zeigen, dass Disstress potentiell zu massiven ‑ auch langfristigen ‑ Veränderungen und Schädigungen stressregulierender Hirnareale führt mit der Folge von Stressregulationsstörungen.
Störungen der Stressregulation können sich individuell unterschiedlich bemerkbar machen, beispielsweise durch eine veränderte Stresstoleranz. Hierbei könnte das Stresssystem schon bei unterschwelligen Ereignissen aktiv werden und massiv Stresshormone ausschütten. Folge: Der Betroffene reagiert schon bei kleineren Anlässen gestresst. Oder: Das Stresssystem bleibt wesentlich länger oder sogar dauerhaft aktiv, obwohl der Stressauslöser längst verschwunden ist oder keine Rolle mehr spielen sollte.
Im Zusammenhang mit affektiven Erkrankungen ist aber wesentlich, dass die stressregulierenden Hirnareale gemäß Gehirnlandkarte auch Affekte steuern und regulieren. Daher besteht die Möglichkeit, dass Disstress neben Stressregulationsstörungen auch direkt affektive Erkrankungen auslöst oder deren Entstehung zumindest begünstigt.
Das bedeutet: Sowohl zwischen Stressor und Stressregulationsstörungen einerseits als auch zwischen Stressor und Affektstörungen andererseits kann ein Kausalzusammenhang bestehen:
Stressauslöser (Stressor) → Disstress → Überforderung stressregulierender Areale → Schädigung stressregulierender Areale → Stressregulationsstörungen
Stressauslöser (Stressor) → Disstress → Überforderung stressregulierender Areale, die auch Affekte steuern → Affektstörungen
Dass einige Hirnareale sowohl für Körperfunktionen ‑ hier: Stressregulation ‑ als auch für die Verarbeitung von Emotionen und Affekten maßgeblich sind, wurde schon im ersten Kapitel thematisiert (→ Abschnitt 1.5 und Abbildungen 5a/5b). Die folgende Abbildung 39 visualisiert diesen Zusammenhang auf der Grundlage von Abbildung 5b als Alternative zur Abbildung 38.
ABBILDUNG 39: STRESSREGULIERENDE UND AFFEKTREGULIERENDE AREALE ALS SCHNITTMENGE
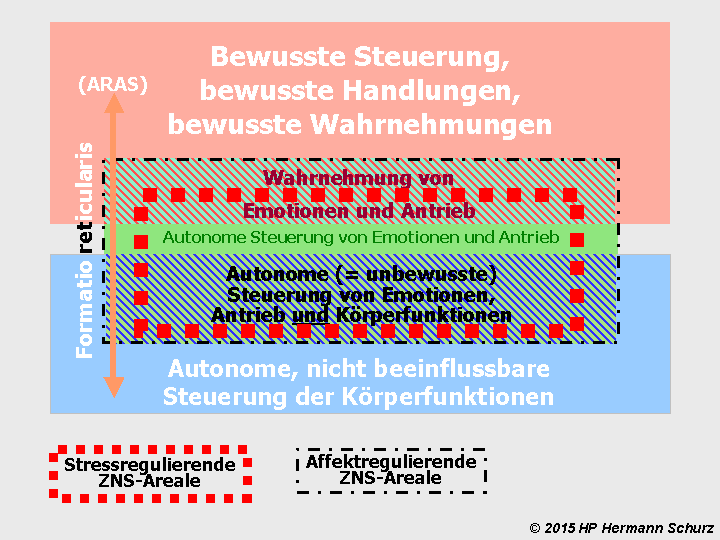
Abbildung 39: Die rot umrandeten funktionalen Bereiche des Gehirns betreffen sowohl Stress- als auch Affektverarbeitung. Schädigt Stress diesen Bereich, ist neben der Stressverarbeitung auch die Affektverarbeitung potentiell negativ betroffen.
Dass Stress grundsätzlich zu Stressregulationsproblemen bzw. Affektstörungen führt, lässt sich aus dem Modell allerdings nicht ableiten. Auch folgende Szenarien sind kausaltheoretisch begründbar:
- Nach einer Stressbelastung treten ausschließlich Stressregulationsstörungen ohne Affektstörungen auf. Der Grund liegt in den individuellen Schädigungen, die durch Stress im Gehirn verursacht werden und die bei jedem Betroffenen anders sein können. Eventuell kommen Affektstörungen im Laufe der Zeit durch eine weitere kontinuierliche Stressschädigung hinzu, das muss aber nicht zwingend so sein.
- Nach einer Stressbelastung treten Affektstörungen ohne Stressregulationsstörungen auf. Die Begründung ist vom Prinzip her mit der vorhergehenden identisch, nur unter umgedrehten Vorzeichen
- Nach einer Stressbelastung treten weder Affektstörungen noch Stressregulationsstörungen auf. Dabei ist es irrelevant, ob der Stress schon zu neurologischen Veränderungen geführt hat ‑ entscheidend ist, ob diese Veränderungen ausreichen, spürbare Symptome zu verursachen.
- Bei allen drei Beispielen kommt es u. a. auf die Widerstandsfähigkeit und den Zustand der stress‑ und affektverarbeitenden Hirnareale an, beispielsweise ob Vorschädigungen bestehen oder nicht. All dies spiegelt damit auch die Stressresilienz Betroffener wider.
Korrelative Zusammenhänge zwischen Stressregulations- und Affektstörungen
Löst Disstress Stressregulations- und Affektstörungen parallel aus, sind diese nicht kausal, sondern korrelativ miteinander verbunden, denn beide Störungen gehen auf eine gemeinsame Ursache zurück, nämlich die stressbedingte physische Schädigung derselben Hirnareale.
Sonstige Auslöser von Stressregulations- und Affektstörungen
Die bisher gemachten Aussagen treffen prinzipiell auch zu, wenn andere Noxen als Disstress die Hirnareale schädigen, beispielsweise Gifte, Sauerstoffmangel oder ein Apoplex (Schlaganfall), denn auch daraus können sowohl Stressregulationsstörungen als auch Affektstörungen resultieren.
Depressive Verstimmungen und Depression als Verstärker
Depressive Verstimmungen und Depression stellen ‑ wie jede schwerwiegende Belastung oder Erkrankung ‑ eigenständige Disstressoren dar (→ Tabelle 19). Insbesondere langfristige depressive Verstimmungen können die stress- bzw. affektrelevanten Areale des Gehirns über die bekannten Stressmechanismen zusätzlich schädigen.
Daher hätten depressive Verstimmungen ebenfalls das Potential, in der Entstehung von Stressregulationsstörungen als auch Affekterkrankungen zu münden: Über diesen Mechanismus kann eine depressive Verstimmung zu einer (klinischen) Depression führen.
Eine schon bestehende (klinische) Depression, die langfristig den Stresslevel erhöht, könnte über diesen Mechanismus sowohl Stressregulationsstörungen begünstigen, aber auch die Depression weiter verstärken.
Die Konsequenzen der Hippocampus-Atrophie nach Stressbelastung für die Neuroplastizität
Die Hippocampusregion gilt innerhalb der affektrelevanten Areale als diejenige, die sich unter Stress am massivsten pathologisch verändert. Häufig werden nach einer starken oder anhaltenden Stressbelastung Atrophien der Hippocampusregion festgestellt, beispielsweise im Zusammenhang mit einer posttraumatischen Belastungsstörung.
Eine Hippocampus-Atrophie hat für das gesamte Gehirn aller Wahrscheinlichkeit nach fatale Folgen, denn der Hippocampus ist sehr stark in die Neurogenese involviert. Im Zuge der Neurogenese reifen u. a. in der Hippocampusregion neue Nervenzellen heran, die später in Hirnregionen wandern, in denen sie benötigt werden. Eine Schädigung der Hippocampusregion könnte daher immer auch eine Behinderung der Neurogenese bedeuten.
In einem solchen Falle könnte das Gehirn nicht mehr in der Lage sein, seine natürliche Fähigkeit zur Neuroplastizität aufrechtzuerhalten oder diese ist zumindest bedeutend erschwert. Dies hätte potentiell zur Folge, dass das Gehirn für viele Erkrankungen anfälliger werden könnte, vor allem für neurodegenerative Erkrankungen, wie Demenz oder Morbus Parkinson oder eine chronische Depression, die Dysthymie oder chronische Stressregulationsstörungen.
4.12.4 Besondere Aspekte psychosozialen Disstresses in Kindheit und Jugend
Durch neuere und exaktere bildgebende Untersuchungsverfahren ‑ wie MRT oder fMRT ‑ hat sich das Verständnis der Gehirnentwicklung von Kindern und Jugendlichen völlig verändert. Waren Wissenschaftler früher davon überzeugt, die Entwicklung des Zentralnervensystems sei ungefähr mit dem 12. Lebensjahr abgeschlossen, geht man nun davon aus, dass das Gehirn bis zum 20. bzw. teilweise auch bis zum 25. Lebensjahr einen komplizierten und langwierigen Aufbau- und Umbauprozess durchmacht. Auch das Wissen über die Komplexität der Gehirnentwicklung Heranwachsender ist durch diesen Fortschritt differenzierter als früher.
Besondere psychosoziale Disstressoren bei Heranwachsenden
Viele der in Tabelle 19 aufgezählten psychosozialen Stressoren (→ Abschnitt 4.12.1) sind auch für Kinder und Jugendliche relevant. In der nachfolgenden Übersicht der Tabelle 21 werden zusätzliche Stressoren aufgezählt, die besonders bzw. ausschließlich in frühen Lebensphasen von Bedeutung sind.
TABELLE 21: BEISPIELE PSYCHOSOZIALEN STRESSES IN KINDHEIT UND JUGEND
Erziehung + Familiäres Umfeld |
Ausbildung |
Allgemein + Sonstiges soziales Umfeld |
|---|---|---|
| Erziehung - Emotionale Misshandlungen - Ständige Entwertungen - Verbale Beschimpfungen - Körperliche Misshandlungen - Überforderung durch die Eltern - Distanzierte Eltern - Desinteressierte Eltern - Elternlosigkeit - Mutterverlust - Längere Trennung von Eltern - Wechsel der Bezugsperson(en) - Frühe Rollenumkehr - Vernachlässigung - Verwahrlosung Sonstige familiäre Disstressoren - Konflikte zwischen den Eltern - Gewalttätigkeit gegen die Mutter - Chronische familiäre Disharmonie - Trennung/Scheidung der Eltern - Geschwisterzuwachs - Auszug aus dem Elternhaus - Sexueller Missbrauch - Kranke/Suchtkranke Eltern - Psychisch kranke Eltern - Längere Abwesenheit v. d. Familie - Häufige Umzüge in der Kindheit |
Kindergarten/Schule - Einschulung - Schulische Überforderung - Schulische Unterforderung - Leistungsdruck - Prüfungsängste - Negative Prüfungserfahrung - Schulabschluss - Entwertung durch Lehrer/Erzieher - Probleme mit Lehrern/Erziehern - Probleme i. d. Klassengemeinschaft - Mobbing - Sexueller Missbrauch |
Soziale Konflikte - Kontaktprobleme mit Gleichaltrigen - Ausgrenzung/Mobbing - Umgang mit Sexualität - Sexuelle Normabweichung - Sexueller Missbrauch - Niedriger sozialökonomischer Status |
Tabelle 21: Diese beispielhafte Aufzählung ist eine Ergänzung der Tabelle 19. Ein besonders schwerwiegendes Problem für Kinder und Jugendliche ist das Aufwachsen in dysfunktionalen Familienverhältnissen, deren Merkmale breit gefächert sind und die nicht nur besonders schwere Fälle, beispielsweise Verwahrlosung oder extreme Formen der Misshandlung, umfassen.
Gehirnentwicklung von der Geburt bis zum 25. Lebensjahr
Anhand gesicherter neuer Erkenntnisse über die Gehirnentwicklung in den ersten 25 Lebensjahren wird verständlich, warum Kindheit und Jugend für die Entstehung psychiatrischer Erkrankungen ‑ auch in späteren Lebensabschnitten ‑ entscheidend sind.
In der gesamten Entwicklungsperiode sind verschiedene Zeitpunkte bzw. Phasen zu unterscheiden, da sie jeweils Entwicklungen einleiten oder beenden: Geburt, 4. Lebensmonat, 10. Lebensmonat, 2. Lebensjahr, 3. Lebensjahr, 4. Lebensjahr, 8. Lebensjahr, 10. Lebensjahr, 14. Lebensjahr, 20. ‑ 25. Lebensjahr und > 25. Lebensjahr.
- Zeitpunkt der Geburt
Bei Geburt beträgt das Gehirngewicht nur ein Viertel des erwachsenen Gehirns. Die Gründe liegen vor allem in den noch sehr kleinen Neuronen, deren unzureichender Vernetzung und einer geringen Myelinisierung der Axone (weiße Substanz). Bei der Geburt liegt also noch ein sehr grober Schaltplan des Gehirns und eine geringe Nervenleitgeschwindigkeit aufgrund unzureichender Myelinisierung vor. Dabei beträgt die Anzahl der Nervenzellen zu diesem Zeitpunkt ungefähr 100 Milliarden. Diese Menge bleibt ‑ bei leichter Abnahme ‑ bis zum Lebensende annähernd konstant.
Unmittelbar nach der Geburt werden im Überfluss neue Verbindungen (Synapsen) zwischen den Nervenzellen aufgebaut. Später werden nicht benötigte Verbindungen nach und nach durch das sogenannte Pruning reduziert und aussortiert. Das geschieht in den drei Cortex-Bereichen somatisch-sensorischer Cortex, parietal-temporaler Cortex und präfrontaler Cortex und den affektrelevanten Hirnarealen mit jeweils unterschiedlicher Geschwindigkeit.
Die Myelinisierung der Axone verstärkt sich mit der Geburt und die Nervenleitungen (Axone) werden dicker und schneller. Dieser Prozess setzt sich bis zum 25. Lebensjahr kontinuierlich fort. Dabei vollzieht sich dieser Prozess nicht gleichmäßig, bestimmte Areale werden zu bestimmten Perioden myelinisiert. Für den Hirnforscher Gerhard Roth betrifft das für die Zeit um die Geburt vor allem die sekundären sensorisch-motorischen Areale (Quelle: Gerhard Roth, Die Entwicklung des kindlichen Gehirns - Normalität und traumatische Störungen, 2011, Institut für Hirnforschung der Universität Bremen).
- 4. Lebensmonat
Relativ schnell hat nach nur vier Monaten die Neuronenverknüpfung im somatosensorischen Cortex ihren Höhepunkt erreicht, ab jetzt wird dort die Vernetzung wieder abgebaut. Die Myelinisierung der assoziativ-okzipitalen, parietalen und temporalen Areale setzt etwa einen Monat nach der Geburt ein und ist im 4. Lebensmonat abgeschlossen.
Es gilt grundsätzlich für diesen wie für alle ähnlichen Prozesse: Je mehr Stimulation das sich vernetzende Gehirnareal erhält, desto mehr Verbindungen können bestehen bleiben. Im angelsächsischen Sprachraum wird hierfür die Redewendung „Use it or lose it“ verwendet. Dabei spielen persönliche Erfahrungen des Kindes und vor allem die elterliche Fürsorge eine erhebliche Rolle.
- 10. Lebensmonat
Nach zehn Monaten ist der Höhepunkt der Vernetzung des parietalen und temporalen Cortex erreicht. Auch hier wird ab jetzt die Vernetzung unter dem Aspekt „Use it or lose it“ abgebaut.
- 2. Lebensjahr
Die Anzahl der Synapsen ist in dieser Zeit etwa identisch mit der im erwachsenen Gehirn
- 3. Lebensjahr
Mit 3 Jahren hat das kindliche Gehirn doppelt so viele Verknüpfungen und den doppelten Energieverbrauch als das eines Erwachsenen, seine Leitungsgeschwindigkeit ist aufgrund der noch unvollständigen Myelinisierung jedoch etwa 16fach geringer.
- 4. Lebensjahr
Abschluss der Vernetzungsphase des somatosensorischen Cortex.
- 8. Lebensjahr
Höhepunkt der Vernetzung und Myelinisierung des präfrontalen Cortex, ab jetzt wird diese Vernetzung wieder abgebaut („Use it or lose it“).
- 10. Lebensjahr
Das 10. Lebensjahr leitet die Adoleszenz-Phase und den Beginn der Pubertät ein. Die Nervenzellenanzahl sinkt jetzt langsam mit einer Rate von 0,7% pro Jahr. Dieser Abbauprozess endet mit dem Abschluss der Adoleszenz-Phase zwischen dem 20. und 25. Lebensjahr.
Der Beginn der Pubertät fällt zusammen mit dem Beginn einer Hochaktivphase der Amygdala und einer Umbauphase affektrelevanter Hirnareale. Die Myelinisierung der orbitofrontalen Areale erfolgt ebenfalls in der Pubertät und setzt sich danach noch einige Zeit fort.
- 14. Lebensjahr
Das Ende der Pubertät fällt zusammen mit dem Abschluss der Vernetzungsphase des parietalen und temporalen Cortex. Bei Mädchen stoppt zusätzlich die Hochaktivphase der Amygdala.
- 20. bis 25. Lebensjahr
In diesem Zeitraum enden mehrere neuronale Entwicklungsprozesse:
- Der Umbau affektrelevanter Hirnareale wird abgeschlossen.
- Bei Männern endet die Hochaktivphase der Amygdala.
- Im Schnitt ist mit dem 22. Lebensjahr auch die Vernetzung des präfrontalen Cortex abgeschlossen.
- Mit dem Abschluss der Myelinisierung wird eine von Geburt an kontinuierlich durchgeführte Entwicklung beendet, die erheblich zur Leistungssteigerung des Gehirns beigetragen hat.
- Ab dem 25. Lebensjahr
Die Neuroplastizität bleibt erhalten, so dass auch im erwachsenen Gehirn eine Entwicklung stattfinden kann. Hier hat der Hippocampus eine wichtig Funktion: Neue Zellen dieser Gehirnregion wurden in anderen Arealen nachgewiesen, so dass von Wanderbewegungen neu entstandener Nervenzellen auszugehen ist.
Hirnentwicklung nach Gerhard Roth
Der Hirnforscher Gerhard Roth teilt die Entwicklung der affektrelevanten Hirnregionen in drei Phasen ein (Quelle: Gerhard Roth, wie oben), die nicht scharf voneinander abzugrenzen sind. Roth verwendet den Fachbegriff der Limbischen Ebene, deren Areale ungefähr mit denen der emotional-affektrelevanten Areale der Gehirnlandkarte übereinstimmen.
- Pränatal: Entwicklung der unteren limbischen Ebene
Entwicklung von Hypothalamus, zentraler Amygdala und Bereichen des Hirnstamms, die das Temperament des Individuums bestimmen sollen und genetischen bzw. vorgeburtlichen Einflüssen unterliegen.
- In den ersten Lebensjahren: Entwicklung der mittleren limbischen Ebene
Entwicklung der basolateralen Amygdala und des mesolimbischen Systems (dazu gehören hauptsächlich der Nucleus accumbens der Basalganglien, Hippocampus und Gyrus cinguli), mit denen der Persönlichkeitskern des Individuums festgelegt sein soll.
- In der späten Kindheit und Jugend: Entwicklung der oberen limbischen Ebene
Roth gibt hier den präfrontalen, orbitofronalen, cingulären und insulären Cortex an mit der Festlegung sozial relevanter Persönlichkeitsmerkmale: Machtstreben, Dominanz, Empathie, die Fähigkeit, Ziele zu verfolgen und Kommunikationsbereitschaft.
Entwickeln sich Stresssysteme hauptsächlich in Kindheit und Jugend?
Es wird noch nicht im Detail verstanden, ob und in welchem Verhältnis sich die verschiedenen Stressachsen in Kindheit und Jugend im Vergleich mit späteren Lebensphasen unterschiedlich entwickeln bzw. verändern.
Wie schon in Abschnitt 4.12.3 gezeigt ist davon auszugehen, dass sich sämtliche Bestandteile des Stresssystems in allen Lebensphasen aufgrund psychosozialer Stresseinwirkung noch verändern (→ Tabelle 20).
Trotzdem scheinen Kindheit und Jugend aufgrund der Erkenntnisse über die Hirnentwicklung besonders relevant zu sein (Quelle: Matz, Pietrek, Rockstroh, Stress in der Kindheit sensitiviert für Stress im Erwachsenenalter - Eine Studie mit psychiatrischen Patienten, 2010, aus: Zeitschrift für Klinische Psychologie und Psychotherapie 39/2010. S. 45 ‑ 55).
Nach Gerhard Roth entsteht das Stresssystem, beispielsweise die für die Cortisol-Ausschüttung zuständige Stressachse, vorgeburtlich bzw. in den ersten Monaten nach der Geburt. So würde die Cortisolrezeptorenanzahl, insbesondere in Amygdala und Hippocampus, in diesem Zeitraum festgelegt (Quelle: Gerhard Roth, wie oben).
ABBILDUNG 40: DIE ENTWICKLUNG DES GEHIRNS IN KINDHEIT UND JUGEND
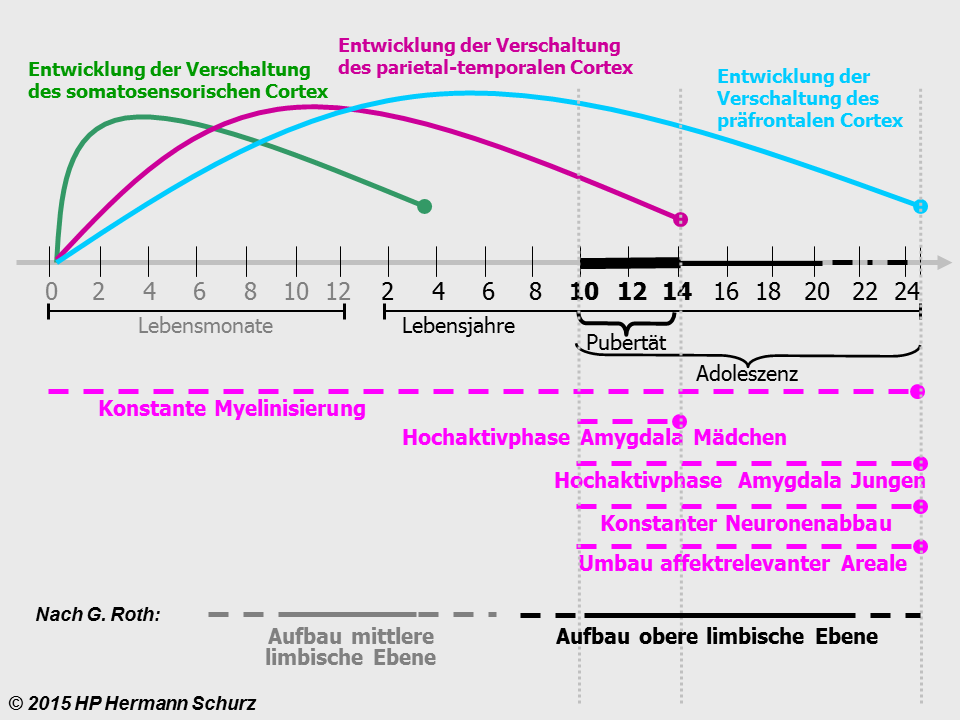
Abbildung 40: Die Entwicklung verschiedener wichtiger Hirnregionen nach der Geburt, während der Pubertät und Adoleszenz. Die gesamte Entwicklung vollzieht sich im Zusammenhang mit der Umgebung des Kindes und seinen Erfahrungen. Unter anderem können chronischer psychosozialer Stress oder auch kurze traumatische Erfahrungen diese Entwicklung nachhaltig stören und führen potentiell zu schweren Hirnentwicklungsstörungen mit Konsequenzen für das gesamte weitere Leben. Nach G. Roth erfolgt der Aufbau der unteren limbischen Ebene pränatal und ist in dieser Graphik nicht berücksichtigt.
Hat psychosozialer Disstress in Kindheit und Jugend besonders gravierende Konsequenzen?
Dass Disstress bei Kindern und Jugendlichen im Vergleich mit Erwachsenen wesentlich gravierendere Folgen haben kann, ist nach den obigen Ausführungen intuitiv verständlich und lässt sich mit den Ergebnissen der Hirnforschung nun auch neurologisch belegen.
Die Hirnentwicklung der ersten 20 bis 25 Lebensjahre stellt eine gewaltige Auf- und Umbauphase dar, und jedwede Störung beeinflusst diesen komplexen Prozess. Stress in Kindheit und Jugend kann daher nicht nur akute Folgen für Heranwachsende haben, auch Hirnentwicklungsstörungen mit Auswirkungen auf sämtliche späteren Lebensphasen scheinen wahrscheinlich.
4.12.5. Empirische Forschung: Zusammenhänge zwischen Stress und Affektstörungen
Ein Kausalzusammenhang zwischen Stress als verursachendem externen Faktor und affektiven Erkrankungen gilt durch die Ergebnisse zahlreicher Untersuchungen als hochwahrscheinlich (Quelle: U. Hapke, U. E. Maske et al., Chronischer Stress bei Erwachsenen in Deutschland, Bundesgesundheitsblatt 2013 (56), S. 769 - 754, Springer-Verlag, Berlin/Heidelberg 2013, http://edoc.rki.de/oa/...pdf).
Studien weisen Zusammenhänge zwischen pathologisch veränderten Stresssystemen und Affektstörungen nach. Die Berliner Charité bietet eine Übersicht über verschiedene Forschungsarbeiten, wobei diese nur einen sehr kleinen Ausschnitt aus der Fülle der Veröffentlichungen darstellt (Quelle: Affektive Störungen und Stresserkrankungen, Klinik für Psychiatrie und Psychotherapie der Berliner Universitätsklinik Charité, https://psychiatrie.charite.de/...).
Folgende in Deutschland durchgeführte Studien bzw. veröffentlichte Fachbeiträge beschäftigten sich mit Zusammenhängen zwischen Stressbelastungen und Affektstörungen:
- Corinna Reck, M. Backenstraß, K. Kronmüller et al., Kritische Lebensereignisse im 2-Jahresverlauf der „Major Depression“ ‑ Eine prospektive Studie mit stationär behandelten Patienten, aus: Nervenarzt 1999, 70, S. 637 ‑ 644, Springer-Verlag GmbH, Berlin/Heidelberg 1999, https://www.researchgate.net/...
In dieser Studie wurde die Bedeutung kritischer Lebensereignisse für den Krankheitsverlauf im Hinblick auf einen Rückfall bei stationär behandelten Patienten mit einer schweren endogenen Depression im Zeitraum von zwei Jahren untersucht. Ein Vergleich mit gesunden Probanden wurde durchgeführt. Patienten mit einer schweren endogenen Depression wiesen sechs bzw. drei Monate vor einer erneuten Klinikeinweisung deutlich mehr negativ bewertete Lebensereignisse auf als Patienten mit einem günstigeren Erkrankungsverlauf ohne Rückfall. Bezogen auf einen Zeitraum von nur drei Monaten vor dem Rückfall hatten die Erkrankten mehr unerwünschte und weniger erwünschte Lebensereignisse als die gesunden Probanden.
- M. Merbach, U. Wittig, E. Brähler, Angst und Depression polnischer und vietnamesischer MigrantInnen in Leipzig unter besonderer Berücksichtigung ihres Eingliederungsprozesses, Medizinische Fakultät der Universität Leipzig, Abteilung für Medizinische Psychologie und Medizinische Soziologie, aus: PPmP Psychotherapie, Psychosomatik, Medizinische Psychologie, 2008, 58 (3/04), S. 146 - 154, Georg Thieme Verlag, Stuttgart/New York, 2008, https://www.researchgate.net/publication/...
Zusammenhänge zwischen Stressbelastung von Migrantinnen und Migranten durch migrationsbedingte Faktoren und deren psychischem Gesundheitszustand war das Thema dieser Untersuchung an der Universität Leipzig. Insbesondere versuchten die Autoren der Studie herauszufinden, welche Migrationsbedingungen im Einzelnen die psychische Gesundheit der Menschen gefährden. Dabei kommentierten die Autoren auch die Ergebnisse vieler früherer Studien, die sich weitgehend mit dem gleichen Thema beschäftigten und bei denen negative Auswirkungen der Migration auf die Psyche festgestellt wurden. Das wichtigste Ergebnis dieser Arbeit war die Feststellung, dass hauptsächlich die soziale Integration und vor allem die individuell wahrgenommene Diskriminierung die Ursachen für Angst- und Depressionserkrankungen waren. Unter sozialer Integration wurde dabei die soziale Unterstützung für den Einzelnen und dessen soziales Netz(‑werk) verstanden. Bei Geschlecht, Sprachkenntnissen bzw. anderen die Qualität der formalen (z. B. kognitiven oder strukturellen) Assimilation betreffenden Merkmalen konnten demgegenüber keine signifikanten Korrelationen mit dem psychischen Gesundheitszustand festgestellt werden bzw. sie waren instabil. Die Ergebnisse waren innerhalb verschiedener Migrantengruppen nicht immer vergleichbar.
- U. Hapke, U. E. Maske, C. Scheidt-Nave, L. Bode, R. Schlack, M. A. Busch, Chronischer Stress bei Erwachsenen in Deutschland, Abteilung für Epidemiologie und Gesundheitsmonitoring, Robert-Koch-Institut, Berlin, veröffentlicht in: Bundesgesundheitsblatt 2013 (56), S. 769 - 754, Springer-Verlag, Berlin/Heidelberg 2013, http://edoc.rki.de/oa/...pdf
Zwischen 2008 und 2011 wurden 8.152 Personen über ihre Stressbelastung, ihren sozioökonomischen Status und die psychische Gesundheit einschließlich Schlafstörungen befragt. Es wurde festgestellt, dass Stressbelastung und sozioökonomischer Status negativ miteinander korrelieren, das heißt je niedriger der Status, desto höher die Stressbelastung. Ähnlich verhält es sich mit dem Zusammenhang zwischen subjektiv wahrgenommener sozialer Unterstützung im privaten Umfeld und Stressbelastung: Je geringer die Unterstützung, desto höher die Stressbelastung. Am wichtigsten sind die Ergebnisse hinsichtlich des psychischen Gesundheitszustandes der Befragten. Stressbelastete Menschen zeigten signifikant häufiger ein Burnout-Syndrom oder eine depressive Symptomatik. Auch Schlafstörungen kamen in diesem Personenkreis häufiger vor.
Die Dortmunder Bundesanstalt für Arbeitsschutz und Arbeitsmedizin untersucht Zusammenhänge zwischen arbeitsbedingtem Stress und Affektiven Störungen:
- Arbeitsbedingtheit depressiver Störungen - Zur Bedeutung arbeitsbedingter Faktoren für das Auftreten depressiver Störungen, Workshop vom 1. Juli 2004 in Berlin, Schriftenreihe der Bundesanstalt für Arbeitsschutz und Arbeitsmedizin, Dortmund, Tagungsbericht 138, Dortmund/Berlin/Dresden 2005, http://www.baua.de/de/...
Spezialisten verschiedener Fachdisziplinen referierten und diskutierten im Rahmen dieser Veranstaltung wissenschaftliche Erkenntnisse zur Frage, ob Affektstörungen bzw. Erkrankungen an einer Depression in einem Zusammenhang mit der Arbeitstätigkeit stehen, das heißt arbeitsassoziiert oder unmittelbar arbeitsbedingt sind, oder ob sie lediglich im Kontext mit der Arbeit auftreten. Dabei wurden Ergebnisse aus verschiedenen Untersuchungen vorgestellt und erörtert. So weist eine Untersuchung deutliche Zusammenhänge zwischen der erhöhten Prävalenz psychischer Störungen und Arbeitslosigkeit im Vergleich mit Erwerbstätigen nach (Rose, 2003). J. Siegrist und A. Rödel vom Institut für Medizinische Soziologie der Heinrich-Heine-Universität Düsseldorf geben einen Überblick über die Ergebnisse empirischer Arbeitsstressforschungen, insbesondere solcher, die sich auf eines der beiden Arbeitsstressmodelle Anforderungs‑Kontroll‑Modell und Modell beruflicher Gratifikationskrisen beziehen. Nach beiden Modellen besteht ein signifikanter Zusammenhang zwischen negativem Arbeitsstress und einer Depression, ebenfalls verbunden mit dem hohen Risiko einer kardio-vaskulären Erkrankung. Auch Renate Rau vom Institut für Organisations- und Sozialpsychologie der Technischen Universität Dresden gibt einen umfangreichen Überblick über verschiedene Untersuchungen der Zusammenhänge zwischen Arbeit und Depression. Dabei weist sie auf die Schwierigkeiten hin, genaue Ursache-Wirkungs-Zusammenhänge zu verstehen und nachzuweisen, ebenso auf die Problematik der Verarbeitung subjektiver Einschätzungen von Befragten, die Arbeitsstudien meist zugrunde liegen. Letzteres betrifft die Einschätzungen und Bewertungen von Arbeit und Arbeitsbelastungen durch die Studienteilnehmer. Aber auch die Erfassung der Depression als Zielvariablen würde in den angeführten Studien völlig unterschiedlich gehandhabt, was eine Vergleichbarkeit erschwere. Das leitet zu einem Referat der Arbeitspsychologin Gisela Mohr über, die einen generellen Überblick über die Erforschung des Zusammenhangs zwischen Erwerbsarbeit und Depressivität aus arbeitspsychologischer Sicht gibt. Sie verweist darauf, dass sie schon 1986 in einer Untersuchung über Stress am Arbeitsplatz einen signifikanten Zusammenhang zwischen depressiven Symptomen und Arbeitsunfähigkeit nachgewiesen hatte. G. Mohr erwähnt darüber hinaus acht Längsschnittstudien, in denen ein Kausalzusammenhang zwischen kritischen Arbeitsbedingungen und Depressivität nachgewiesen wurde. Die beiden Organisationspsychologen Diether Gebert und Lutz von Rosenstiel kämen nach einer Auswertung von Längsschnittstudien zu dem Schluss, dass Arbeitsstress sogar ein wesentlich stärkerer Stressor für die Entwicklung von Depressivität sei als der familiäre Stress, so Gisela Mohr.
- R. Rau, N. Gebele, K. Morling, U. Rösler, Untersuchung arbeitsbedingter Ursachen für das Auftreten von depressiven Störungen, Projekt F 1865, Philipps-Universität Marburg, Fachbereich Psychologie/Arbeits- und Organisationspsychologie und Bundesanstalt für Arbeitsschutz und Arbeitsmedizin, Dortmund, Dortmund/Berlin/Dresden 2010, http://www.baua.de/de/Publikationen/...
Neben einer Beantwortung von Fragen arbeitsbedingter Ursachen für das Auftreten von Depressivität (Major Depression oder depressive Verstimmungen) sollte in dieser Studie vor allem geprüft werden, ob eine im Vergleich mit früheren Erhebungsmethoden als objektiver eingeschätzte Vorgehensweise ebenfalls Zusammenhänge zwischen bestimmten Merkmalen der Arbeitstätigkeit und dem Auftreten von Depressionen nachzuweisen in der Lage ist. Kritisiert wurde nämlich, dass in fast allen bis dahin durchführten Studien jeweils dieselben Personen sowohl über ihre Bewertung von Arbeitsmerkmalen als auch zur Depressivität befragt wurden. Man vermutete, dass es dadurch zu falschen oder zumindest übetriebenen Darstellungen gekommen sei. Daher sollte nun geprüft werden, zu welchen Ergebnissen unabhängig von den Arbeitsplatzinhabern erfasste Bewertungen der Arbeitsmerkmale führen. Mit dieser Studie konnte unter dem veränderten Studien-Design erneut belegt werden, dass auch die (nun objektiv) bewertete Arbeitsintensität mit dem Auftreten einer Depression im Zusammenhang steht. Das heißt auch, dass Erfasssungen subjektiver Wahrnehmungen in früheren Studien nicht automatisch zu verzerrten oder sogar falschen Ergebnissen geführt haben. Darüber hinaus belegte diese Studie, dass ein objektiv bewerteter Tätigkeitsspielraum mit dem Auftreten einer Depression in keinem Zusammenhang steht. In den früheren Studien korrelierte ein subjektiv bewerteter geringer Tätigkeitsspielraum häufig mit dem Auftreten von Depressivität. Das heißt: Je geringer der (subjektiv empfundene) Tätigkeitsspielraum, desto eher die Neigung zur Depressivität. Hier führte die veränderten Methode also zu einem gegenteiligen Ergebnis. Es scheint so zu sein, dass Beschäftigte mit einer Depression krankheitsbedingt ihren Tätigkeitsspielraum subjektiv als zu gering einschätzen. Damit gäbe es zwar einen Zusammenhang zwischen Depression und subjektiv erlebtem Tätigkeitsspielraum, nicht jedoch zwischen Depression und objektiv bewertetem Tätigkeitsspielraum.
Besonders interessant sind Studien, mit denen der Nachweis hirnorganischer Auswirkungen starken Stresses gelingt:
- Eberhard Fuchs, Gabriele Flügge, Psychosoziale Belastung als Ursache molekularer und struktureller Veränderungen im Gehirn - Wie tierexperimentelle Untersuchungen zum Verständnis der Pathomechanismen depressiver Erkrankungen beitragen können, Zeitschrift für Psychosomatische Medizin und Psychologie, 2001, 47/1, S. 80 ‑ 97, Verlag Vandenhoeck & Ruprecht, Göttingen 2001, http://www.vr‑elibrary.de/doi/...
Die Ergebnisse dieser Tierversuchsstudie mit Tupaia belangeri (eine Tierart, die für Untersuchungen neurobiologischer Grundlagen depressiver Erkrankungen geeignet sein soll) zeigen, dass Disstress sowohl zu reversiblen als auch zu langfristigen Veränderungen im Zentralnervensystem führt. Ausgangspunkt waren experimentell herbeigeführte soziale Konfrontationen zwischen jeweils zwei männlichen Tieren über mehrere Wochen, wobei das jeweils unterlegene der beiden Tiere typische Stressreaktionen und chronische psychosoziale Belastungen zeigte. Diese lösten eine Hyperaktivität der Hypothalamus-Hypophysen-Nebennierenrinden- und der Hypothalamus-Sympathikus-Stressachse aus. Die beiden Autoren wiesen nach, dass es beim gestressten Tier zu morphologischen Veränderungen von Nervenzellen des Hippocampus kam, ebenfalls zu Veränderungen der Neurogenese im Hippocampus. Es änderte sich darüber hinaus die Expression (Proteinsynthese) von Glucocorticoid-, serotonergen und noradrenergen Rezeptoren. Die Ausmaße der Veränderungen korrelierten mit der Stressdauer.
4.12.6 Fazit: Psychosozialer Disstress und Affektstörungen
Gefahrensituationen früher
Schon seit Urzeiten lösen Gefahrensituationen bei Menschen und Tieren Angst- und Fluchtreaktionen aus. Sie dienen dazu, sich möglichst schnell in Sicherheit zu bringen und sind wichtige Voraussetzungen, die Überlebenswahrscheinlichkeit Einzelner oder einer Gruppe zu erhöhen. Die physiologischen Voraussetzungen dieser Stressreaktionen werden vom Gehirn zusammen mit peripheren Organen gesteuert.
Typischerweise führten in archaischen Zeiten kurzfristige Gefahrensituationen zu derartigen Stressreaktionen, beispielsweise plötzliche Konfrontationen mit gefährlichen Fressfeinden, sonstigen Gegnern oder Jagdaktivitäten. Alle nervlich-physiologischen Reaktionen waren dementsprechend ebenfalls nur für Sekunden bis wenigen Minuten notwendig und die Regelsysteme konnten den Normalzustand zügig wiederherstellen.
Kurzfristiger Stress und kurzfristige Stressreaktionen sind daher völlig normal. Sowohl Nerven- als auch Hormonsystem sind an derartige Situationen perfekt angepasst.
Gefahrensituationen heute
Aufgrund des gesellschaftlich-kulturellen Wandels haben heutige Gefahrensituationen im Vergleich dazu einen langfristigen Charakter:
- Heutige Gefahren sind gesellschaftlich‑sozialer Natur, beispielsweise (drohende) Arbeitslosigkeit, disharmonische Familienverhältnisse, chronische berufliche Überlastung oder soziale Ausgrenzung.
- Heutztage sind die Möglichkeiten eines kurzfristigen Frustrationsabbaus begrenzt, denn das wird ‑ aus guten Gründen ‑ gesellschaftlich nicht toleriert, führt jedoch tendenziell zur Verlängerung von Stresssituationen.
- Durch die höhere Lebenserwartung im Vergleich zu früher können Folgen negativen Stresses, die sich heutzutage eher über einen längeren Zeitraum entwickeln, überhaupt erst zum Vorschein kommen.
Trotz dieses Wandels sind die menschlichen Stresssysteme dennoch weiterhin darauf ausgelegt, kurzfristigen Stress zu parieren. Es besteht deshalb bei heutigem Langzeitstress eine hohe Gefahr, diese Systeme zu überfordern und pathologisch zu verändern. Neben peripheren Organen - wie Schilddrüsen oder Nebennieren - sind vor allem zentralnervöse Strukturen Bestandteile der Stressysteme und daher von potentieller Erkrankung durch Stress betroffen.
Die Verbindung zwischen Stress und Affektiven Störungen ist gut nachzuvollziehen: Da die stressregulierenden Gehirnareale ebenfalls für die Verarbeitung und Regulierung von Affekten zuständig sind, führt eine stressbedingte Erkrankung dieser Hirnareale mit großer Wahrscheinlcihkeit parallel zu affektiven Erkrankungen.
Aus diesen Gründen ist es weder sinnvoll noch möglich, nach einem bestimmten Prozess und/oder einer bestimmten Substanz zu suchen, die für eine dysfunktionale Stressverarbeitung verantwortlich sein könnten. Vielmehr ist die Erkrankung der Stressverarbeitungssysteme ein ‑ je nach individueller Lage ‑ langsamer oder schneller vonstatten gehender Degenerationsprozess, der das Gesamtsystem oder wesentliche Teile davon betrifft.
Die pathologischen Entwicklungen können verschiedene Abläufe und Substanzen betreffen, die in der Stressverarbeitung relevant sind und im individuellen Einzelfall stark voneinander abweichen und systembedingt schwer ermittelbar sind.
Die Komplexizität der Stressverarbeitung wird anhand einer Liste deutlich, die alle derzeit bekannten beteiligten Organe und körpereigene Substanzen aufzählt und die alle von länger andauerndem psychosozialem Disstress betroffen ‑ und damit gefährdet ‑ sind, darunter auch viele Bereiche des Zentralnervensystems:
- Amygdala
- Formatio reticularis
- Hippocampus
- Hypophyse
- Hypothalamus
- Weitere affektrelevante ZNS-Areale (Basalganglien, Gyrus cinguli, Thalamus etc.)
- Nebennierenmark
- Nebenierenrinde
- Schilddrüse
- Acetylcholin
- Adrenalin
- ANP
- Cortisol
- Noradrenalin
- Als Releasing-Hormone ADH, CRH und TRH, davon ADH und CRH auch als Wirk-Hormone
- Als Steuer-Hormone ACTH und TSH
- Thyroxin
Zusammenhänge zwischen Stress, Stressregulationsstörungen und Affektstörungen
Stressbedingte schädliche physiologische Prozesse führen zu ungünstigen strukturellen bzw. degenerativen Veränderungen in Hirnarealen, die sowohl die Stressverarbeitung als auch die Affektverarbeitung betreffen. Führen diese Veränderungen zur Dysfunktionalität der Areale, besteht damit eine hohe Wahrscheinlichkeit sowohl für Stressregulationsstörungen als auch für Affektstörungen.
Starker und/oder chronischer (Dis‑)Stress löst damit sowohl Stressregulationsstörungen als auch Affektstörungen aus. Damit besteht jeweils ein kausaler Zusammenhang sowohl zwischen (Dis‑)Stress und Stressregulationsstörungen als auch zwischen (Dis‑)Stress und Affektstörungen:
- Disstress → Belastung/Schädigung stressrelevanter Hirnareale → Stressregulationsstörungen
- Disstress → Belastung/Schädigung stressrelevanter (= affektrelevanter) Hirnareale → Affektstörungen
Damit ist ein korrelativer Zusammenhang zwischen Stressregulations- und Affektstörungen begründbar, während ein Kusalzusammenhang zwischen Stressregulationsstörungen und Affektstörungen mit diesem Modell nicht zu belegen ist.
ABBILDUNG 41: DISSTRESS, STRESSREGULATIONSSTÖRUNGEN UND AFFEKTSTÖRUNGEN
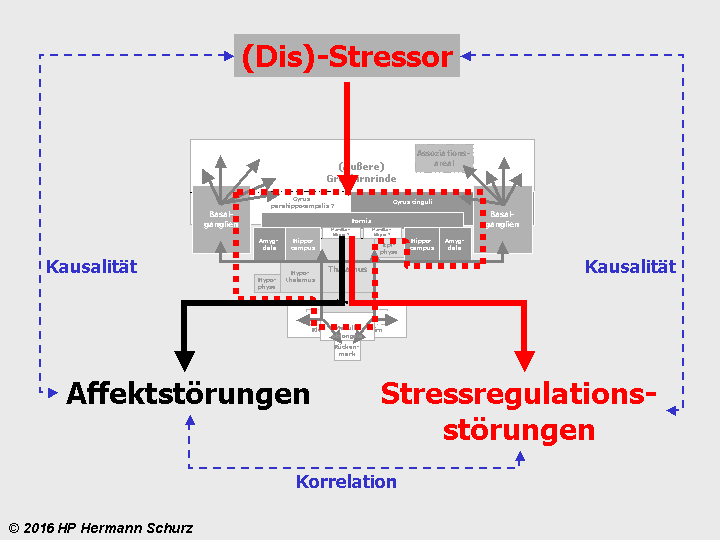
Abbildung 41: Dass es zwischen der krankhaften Veränderung der Stresssysteme und Affektstörungen einen Zusammenhang geben muss, ist schon intuitiv verständlich. Dass dieser Zusammenhang einen kausalen Charakter hat, ist mit den hier zugrunde liegenden Modellen nicht zu begründen. Dass die beiden Zustände Ergebnisse einer gemeinsamen Ursache - nämlich Disstress - sind und damit korrelativ miteinander verbunden sind, ergibt sich unmittelbar aus dem kausaltheoretischen Modell, denn die von starkem Disstress in Mitleidenschaft gezogenen Hirnregionen betreffen zwar in erster Linie stressregulative Hirnregionen, diese sind parallel jedoch auch für die Affektverarbeitung zuständig. Dass nach einer längerfristigen Stressbelastung sowohl Symptome einer aus dem Gleichgewicht geratenen Stressregulation als auch Symptome einer Affekterkrankung auftreten, ist damit hochwahrscheinlch. Eine Kausalität besteht jeweils zwischen Disstress und Affektstörungen bzw. zwischen Disstress und Störungen der Stressregulation.
Auswirkungen von psychosozialem Stress in Kindheit und Jugend
Das Gehirn ist zwischen Geburt und dem 20./25. Lebensjahr mit einer großen Baustelle vergleichbar. Dementsprechend sind die Auswirkungen des psychosozialen Stresses, die schon im Zentralnervensystem eines Erwachsenen großen Schaden anrichten können, in dieser Phase noch gefährlicher.
Neben einer allgemein höheren Empfindlichkeit gegenüber Disstress können zusätzlich Hirnentwicklungsprozesse massiv gestört werden mit weitreichenden Folgen für das gesamte weitere Leben.
Die Ausprägungen der Schädigungen hängen auch von der Entwicklungsphase ab, in der sich das Gehirn zum Zeitpunkt/Zeitraum der Stressphase befindet. Genauere Aussage oder Prognosen zu einer individuellen Situation sind schwierg, wenn nicht sogar ganz unmöglich.
4.13 Sonstige abiotische physikalisch-chemische Noxen ▲
Zur Systematik der Darstellung von Wirkungen sonstiger abiotischer Substanzen bzw. Noxen
Abiotischer Stress resultiert bei allen physikalischen und den meisten chemischen Noxen, beispielsweise Strahlung, Giften oder mechanischen Verletzungen, aus einer meist direkten Wirkung von außen (exogen) auf den Organismus bzw. seine Organe und Zellen. Von derartigem Stress ist das Zentralnervensystem, vor allem das Gehirn, besonders stark gefährdet.
Die Kausalfaktoren Mikronährstoffe, Sauerstoff, Wasser, Lipide und Aminosäuren gehören von ihrer Systematik zwar zu den abiotisch-chemischen Substanzen, die im speziellen Falle einer Unterversorgung Zellstress verursachen. Mangelversorgung und Mängel von Kausalfaktoren sind Thema von Kapitel 4 A. Die Gefahren einer potentiellen Überversorgung mit Kausalfaktoren, beispielsweise Mikronährstoffen, wird im Rahmen verschiedener therapeutischer Ansätze in den Kapiteln 5 und 6 des zweiten Teils diskutiert. Lediglich die Problematik der Sauerstoffüberversorgung, die in bestimmten grenzwertigen Lebenssituationen auftreten kann, wird im Zusammenhang mit den negativen Folgen der endogenen Zellatmung und reaktiver Sauerstoff-Spezies (ROS) in diesem Kapitel erörtert (→ Abschnitt 4.13.6 bzw. den nachfolgenden Unterabschnitt mit einer allgemeinen Übersicht über ROS).
Es sind abiotisch-physikalische Stressoren und abiotisch-chemische Stressoren zu unterscheiden.
Abiotisch-physikalische Stressoren
Verschiedene physikalische Stressoren stehen im Verdacht, den menschlichen Organismus negativ zu beeinflussen und zu schädigen. Sie können mutagen an der DNA wirken, aber auch sonstige Zellbestandteile angreifen, beispielsweise innere oder äußere Zellmembranen. Oft tun sie das durch die Bildung freier Radikaler oder aggressiver reaktiver Sauerstoff-Spezies (ROS). Entscheidend ist, ob das im Zentralnervensystem und insbesondere in seinen affektrelevanten Arealen geschieht. Folgende physikalische Stressoren stehen im Mittelpunkt der Diskussionen nachfolgender Abschnitte:
- Licht-/Tageslichtmangel und Mangel an UV-B-Strahlung (→ Abschnitt 4.13.1)
Bei Lichtstress geht es hauptsächlich um Tageslichtmangel. Unter Umständen kann ein Zuviel an Licht eine Reizüberflutung darstellen und dadurch Bedeutung als psychosozialer Stressor erlangen (→ Abschnitt 4.12), was in Abschnitt 4.13.1 jedoch nicht erörtert wird.
Die Problematik einer sinkenden Vitamin‑D‑Produktion bei Lichtmangel bzw. Mangel an UV‑B‑Strahlung in der Haut mit ihren Auswirkungen auf das Gehirn wird im Zusammenhang mit der saisonal abhängigen Depression (SAD) bzw. der Lichtmangeldepression diskutiert.
- Nicht-ionisierende Mobilfunkstrahlung (→ Abschnitt 4.13.2)
Es ist strittig, ob hochfrequente elektromagnetische Mobilfunkwellen gesundheitsschädigend sind. Es werden thermische und athermische Auswirkungen diskutiert.
- Ionisierende Strahlung (→ Abschnitt 4.13.3)
Ionisierende Strahlung kann sowohl natürlichen als auch künstlichen Urspungs sein und hat ein hohes Potential, Zellbestandteile, insbesondere Zellmembranen und DNA, zu schädigen.
Die umweltbedingten natürlichen radioaktiven Alpha-, Beta- oder Gamma-Strahlen stellen natürlich-induzierte Mutagene dar.
Die zusätzlichen Belastungen durch die technische Nutzung von radioaktivem Material oder dessen Strahlung, beispielsweise in Kraftwerken oder in der nuklearmedizinischen Diagnostik bzw. Therapie, zählen demgegenüber zu den exogen-induzierten Mutagenen.
Künstliche ionisierende Strahlung ist in der Diagnose und Behandlung neurologischer Erkrankungen von Gehirn und Rückenmark ein wichtiges Instrument. Die Strahlen werden hauptsächlich in der Tumor- und Krebstherapie verwendet wird. Auch bei der Röntgen- oder Protonenstrahlung handelt es sich um künstlich erzeugte Strahlungsarten, die hauptsächlich für medizinische Anwendungen genutzt werden.
- Hirnverletzungen (mechanisch bedingte Traumata) (→ Abschnitt 4.13.4)
Mechanisch bedingte Verletzungen des Zentralnervensystems durch Stoß oder Schlag sind häufiger als man denkt und kommen nicht nur bei Boxern, Fußball- oder Eishockeyspielern vor. Sie werden in den Formen der Gehirnerschüttung, des Schädel-Hirn-Traums (SHT) und der Chronisch-traumatischen Enzephalopatie (CTE) diskutiert.
Ebenfalls können Tumore oder überschüssiges Gehirnwasser Ursachen für ein Hirntrauma sein, da sie permanent Druck auf das Hirngewebe ausüben. Derartige medizinisch bedingte Noxen werden jedoch in Abschnitt 14.14 dikutiert.
- Schall/Lärm (→ Abschnitt 4.13.5)
Hier geht es hauptsächlich um ein Zuviel von Schall bzw. Lärm, in Ausnahmefällen kann auch die Abwesenheit von Geräuschen Stress verursachen.
Neben der Schädigung des Hörapparates hat Lärm eine Bedeutung als psychosozialer Disstressor. Es spielen daher Mechanismen eine Rolle, die auch bei psychosozialen Disstressoren relevant sind (→ Abschnitt 4.12).
Abiotisch-chemische Stressoren
Auch chemische Stressoren stehen im Verdacht oder sind nachgewiesenermaßen in der Lage, den menschlichen Organismus bzw. das Zentralnervensystem negativ zu beeinflussen und zu schädigen. Sie können mutagen an der DNA wirken, aber auch sonstige Zellbestandteile angreifen, beispielsweise innere oder äußere Zellmembranen. Chemische Stressoren sind ebenfalls an der Bildung aggressiver reaktiver Sauerstoff-Spezies (ROS) beteiligt.
Die Stresspotentiale folgender chemischer Noxen stehen hier im Mittelpunkt:
- Zellatmung und endogene Energiestoffwechselprozesse (→ Abschnitt 4.13.6)
Die Bildung reaktiver Sauerstoff-Spezies (ROS), welche nicht durch die antioxidativen Prozesse neutralisiert werden können, schädigt Zellgewebe durch Oxidation. ROS entstehen vor allem bei der Zellatmung.
- Exogene Gift- und Schadstoffe ohne Suchtstoffe (→ Abschnitt 4.13.7)
Es werden verschiedene Gift- und Schadstoffe auf ihr nervenschädigendes Potential, insbesondere im Zusammenhang mit Affektstörungen, untersucht.
Es geht um Substanzen, die der Herstellung und Verwendung von Kunststoffen, Farben und Lacken, Kraftstoffen (Benzin, Diesel), Holzschutz-, Pflanzenschutz- und Reinigungsmitteln sowie der Schädlings- bzw. Insektenbekämpfung einschließlich der Pilz- und Sporenbekämpfung (Fungizide) und der Unkrautvernichtung dienen.
Darüber hinaus sind noch Schadstoffe aus Verbrennungsprozessen relevant, wobei Stickstoffdioxid (NO2), Feinstaub (<10 µm Ø), lungengängiger Feinststaub (< 2,5 µm Ø) bzw. ultrafeine Partikel (< 0,1 µm Ø) unterschieden werden.
Die Belastung durch Mikroplastik gerät in letzter Zeit immer mehr in den Fokus der Hirnforscher und Mediziner, da es Anhaltspunkte dafür gibt, dass Mikroplastik die Blut-Hirn-Schranke überwindet.
- Suchtstoffe (→ Abschnitt 4.13.8)
Rauschgifte schädigen zweifelsfrei das Zentralnervensystem. Es werden die wichtigsten Substanzen untersucht: Nikotin/Rauchen, Alkohol, Cannabis/THC, Ecstasy, Crystal Meth, Kokain und Heroin.
Ein weiteres Thema ist die Bewertung der Forderungen nach der Freigabe bestimmter Rauschmittel, beispielsweise Cannabis/THC. Die Problematiken einer Freigabe von THC werden insbesondere unter dem Aspekt der Gehirnentwicklung von Kindern, Jugendlichen und jungen Erwachsenen erörtert.
Exogene abiotische Stressoren und Kausalfaktormangel bzw. Kausalfaktormängel
Einige abitotische Stressoren stehen häufig im Zusammenhang mit Themen aus Kapitel 4 A, da sie zu Kausalfaktormängeln oder Kausalfaktormangel führen, beispielsweise durch die Verursachung von Nukleinsäureschäden, Kausalfaktorfehlversorgungen oder psychosozialem Disstress.
So sind DNA-Mutationen oftmals Folgen ionisierender Strahlen und den damit verbundenen freien Radikalen, die Biomoleküle aufzubrechen in der Lage sind. Lichtmangel hat unter Umständen eine Kausalfaktormangelversorgung als Konsequenz und die Verarbeitung von Sauerstoff zur Energiegewinnung führt unter Umständen zu Zellschäden aufgrund freier Sauerstoffradikaler und hochreaktiver Sauerstoffverbindungen (ROS), ähnlich denen aufgrund ionisierender Strahlung. Psycho‑sozialer Stress kann unter anderem durch Lärm ausgelöst werden und durch die Notwendigkeit des erhöhten Abbaus von Stresshormonen verstärkt hochreaktive Sauerstoff‑Spezies (ROS) zur Folge haben.
Um redundante Beschreibungen zu vermeiden, wird daher bei Bedarf auf ‑ meist vorhergehende ‑ Abschnitte Bezug genommen, in denen grundlegende Schädigungsmechanismen und deren Folgen erörtert werden. Wenn jedoch nötig, werden Themen wiederholt dargestellt.
Freie Radikale und reaktive Sauerstoff-Spezies (ROS) triggern degenerative Prozesse
Einen erheblichen Anteil an der Schädigung von Zellen bzw. Geweben durch exogene Noxen und der damit verbundenen degenerativen Prozesse haben freie Radikale und ROS. Sie entstehen aber auch durch endogene Vorgänge im Zellinneren. Selbst im Zusammenhang mit den oben beschriebenen Auswirkungen psychischen Stresses spielen ROS eine indirekte Rolle, worauf schon verwiesen wurde.
Freie Radikale und ROS sind der Schlüssel zum Verständnis vieler nachfolgend beschriebener Vorgänge und sollen in diesem einführenden Abschnitt daher etwas mehr Aufmerksamkeit erhalten. In der Beschreibung der verschiedenen Noxen (→ Abschnitte 4.13.1 ff.) wird auf deren Eigenschaften, freie Radikale oder ROS zu verursachen, jeweils verwiesen.
Freie Radikale und ROS (ROS für den englischen Fachterminus Reactive Oxygen Spezies, auf Deutsch: reaktive Sauerstoff-Spezies) sind das Ergebnis zahlreicher physikalisch-chemischer Noxen. Sie sind strikt zu unterscheiden und lösen ‑ sofern sie nicht von der Zelle neutralisiert werden ‑ zellschädigende Prozesse aus, die Proteine und die DNA genauso wie Lipide bedrohen ‑ und damit sämtliche Gewebearten einschließlich der wichtigen Zellmembranen. Beide Schadsubstanzen werden oft miteinander verwechselt und ihre Entstehungsprozesse und Folgen nicht ausreichend differenziert, was zur Verwirrung führen kann:
- Direkte freie Radikale entstehen durch verschiedenartige ionisierende Strahlen, zum Beispiel elektromagnetische Strahlen oder Teilchenstrahlung (→ Abschnitt 14.13.3) unmittelbar am Gewebe. Davon sind sämtliche Gewebearten bzw. Zellsubstanzen betroffen, deren Atome und Moleküle auf diese Weise durch ionisierende Strahlung verändert werden können. Dieser Vorgang wird auch Ionisation genannt.
- ROS bestehen aus zwei Gruppen schädlicher aggressiver Substanzen: (1.) den freien Sauerstoffradikalen und (2.) den nicht-radikalen reaktiven Sauerstoffverbindungen. Als ROS werden reaktionsfreudige Verbindungen bezeichnet, die mit Sauerstoff in einem Zusammenhang stehen.
ROS haben eine mit der direkten Ionisation vergleichbare schädliche Wirkung auf das Gewebe, und das ist auch einer der Gründe, warum ROS und direkte freie Radikale oft in einen Topf geworfen werden. Hauptverusacher von ROS sind körper- bzw. zellinnere Prozesse, vor allem Energieversorgungsprozesse, bei denen Sauerstoff naturgemäß eine besonders wichtige Bedeutung hat. Daher werden ROS - abgeleitet vom lateinischen Wort Oxygenimum für Sauerstoff - auch Oxidantien genannt.
ROS entstehen ebenfalls durch ionisierende Strahlen, die Wasser durch Wasserradiolyse verändern. Für eine Zelle, die zu 80% aus Wasser besteht, ist dies nicht minder gefährlich. Damit sind ionisierende Strahlen, neben der direkten Gewebeschädigung durch Radikalbildung mittels Ionisation, auch zu einer indirekten Schädigung des Zellgewebes durch von ihnen verursachte Wasserradiolyse in der Lage.
Es gibt Untersuchungen über das Verhältnis beider Mechanismen bezüglich von ihnen verursachter Schäden. So soll der Anteil direkter Gewebeschädigungen durch Ionisation ca. 30 bis 40% betragen, der Anteil der Schädigungen durch ROS ca. 60 bis 70% (Quelle: Patricia Virsik-Köpp, Biologische Strahlenwirkungen, Zentrum Radiologie, Universitätsmedizin Göttingen, https://www.uni‑goettingen.de/de/document/... ).
Ursachen direkter freier Radikaler und reaktiver Sauerstoff-Spezies (ROS) in der Übersicht
Reaktive Sauerstoff-Spezies (ROS) sind die Folgen vieler zellendogener Prozesse.
- ROS entstehen in Körper- und Nervenzellen aufgrund natürlicher Stoffwechselprozesse:
- Schon der Energiestoffwechsel einer normalen Zellatmung (→ Abschnitt 4.13.6) hat ROS zur Folge.
- Eine verstärkte Zellatmung bei hohem Energiestoffwechsel führt zu einer entsprechend höheren ROS-Produktion (→ Abschnitt 4.13.6).
- ROS entstehen durch das Immunsystem bei der Aktivierung von Granulozyten und Makrophagen.
- Im Zentralnervensystem entstehen ROS bei der Fenton-Reaktion durch nicht proteingebundenes Eisen.
- Beim Hormonabbau, insbesondere beim Abbau von Stresshormonen, entstehen ROS (→ Abschnitt 4.12).
- Die Aktivitäten verschiedener Oxidasen, beispielsweise Monoaminooxidase in Nervenzellen, Xanthinoxidase, L‑Aminosäureoxidase oder Tyrosinhydroxylase, führen zu einer erhöhten Anzahl von ROS-Substanzen.
- ROS werden bei der Verstoffwechslung der Arachindonsäure gebildet.
- Durch externe ionisierende Strahlung (→ Abschnitt 4.13.3) entstehen freie Radikale direkt am Zellgewebe (Ionisation). Ebenfalls sind ROS durch Wasserradiolyse der Zellflüssigkeit die Folgen ionisierender Strahlung. Diese Strahlungsarten sind relevant:
- Natürliche und künstliche UV-A-/UV-B-Strahlung, die ausschließlich die oberen Hautschichten betreffen.
- Natürliche oder künstliche Alpha- und Beta-Strahlung, die ebenfalls nur die oberen Hautschichten und ggf. die Augenoberfläche betreffen.
- Tief in den Körper eindringende künstliche, natürliche oder therapeutisch anzuwendende Gamma-Strahlung.
- Tief in den Körper eindringende künstliche Röntgen- und Protonenstrahlung, beispielsweise im Rahmen einer nuklearmedizinischen Diagnose oder Therapie.
- Auch von außen in der Körper eindringende ionisierende Substanzen oder andere Schadstoffe haben sowohl freie Radikale als auch ROS als Folgen:
- Direkte Ionisation und ROS durch von Nahrung und Atmung aufgenommene Substanzen, die ionisierende Alpha-, Beta- oder Gamma-Strahlen emittieren, auch im Zusammenhang mit einer therapeutischen Anwendung (→ Abschnitt 4.13.3).
- Direkte Ionisation und ROS durch Implantation kleiner Strahlenquellen in den Körper im Rahmen einer nuklerarmedizinischen Brachytherapie (→ Abschnitt 4.13.3).
- Direkte Ionisation und ROS durch Injektionen ionisierender Substanzen im Rahmen einer nuklearmedizinischen Behandlung (→ Abschnitt 4.13.3).
- ROS entstehen auch durch eingeatmeten Tabakrauch und Luftschadstoffe.
- ROS entstehen durch mit der Nahrung aufgenommene Umweltgifte, beispielsweise Chemikalien, Lösungsmittel oder Rückstände von Pflanzenschutzmitteln.
- Aber auch mit der Nahrung aufgenommene Lebensmittelzusatzstoffe führen zu einem vermehrten Aufkommen von ROS.
- Die Einnahme von Medikamenten kann ebenfalls einen Anstieg von ROS zur Folge haben.
Potentielle positive ROS-Eigenschaften
Studien deuten drauf hin, dass die ROS-Substanzen Wasserstoffperoxid und Hyperoxid bei Signalübertragung und Blutgefäßerweiterung (Vasodilatation) im Gehirn eine positive Rolle spielen. Hier ist der Kenntnisstand aber noch gering (Quellen: S. G. Rhee, Redox signaling: hydrogen peroxide as intracellular messenger, 6/1999, EMM Experimental & Molecular Medicine, Nature Publishing Group, London/UK 1999) und K. T. Kishida, E. Klann, Sources and targets of reactive oxygen species in synaptic plasticity and memory, Antioxidants & Redox Signaling, 2/2007, Mary Ann Liebert, Inc., publishers, New Rochelle, New York/USA, 2007). Etwaige stoffwechselrelevante positive Funktionen der ROS sollen jedoch nicht erörtert werden.
4.13.1 Lichtmangel und Affektstörungen ▲
Lichtmangeldepression vs. saisonal abhängige Depression (SAD)
Potentielle Auswirkungen eines Lichtmangels auf Affekte, auch Lichtmangeldepression genannt, sind von der saisonal abhängigen Depression (SAD für Seasonal affective disorder) zu differenzieren. Die Lichtmangeldepression wird fälschlicherweise häufig mit der SAD gleichgesetzt. Auch der missverständliche Begriff „Winterdepression“ ist sehr populär. Als kritische Jahreszeit gilt dementsprechend die lichtarme Zeit zwischen Spätherbst und Frühjahr von etwa Oktober bis einschließlich März. Die SAD berücksichtigt jedoch noch weitere saisonale Phänomene, beispielsweise Temperaturveränderungen, Verkürzungen der Helligkeitsphasen und/oder saisonal bedingte Reduktionen der Aktivitäten bzw. Veränderungen des Biorhythmus. Einige dieser Phänomene können auch im Frühjahr oder Sommer relevant sein.
Die SAD wird in der internationalen Klassifikation ICD‑10 bzw. der aktuellen deutschen Version ICD‑10‑GM Version 2021 lediglich zwei Unterkategorien der rezidivierenden depressiven Störung (F 33) zugeordnet, die sich auf leichtere (F 33.0) bzw. mittelgradige (F 33.1) rezidivierende depressive Störungen beziehen. Nähere Erklärungen gibt es dazu nicht (Quelle: Bundesinstitut für Arzeimittel und Medizinprodukte, www.dimdi.de/static/...). Die SAD gilt damit nicht als eigenständige Erkrankung. Auch im aktuellen US‑amerikanischen Pendant DSM‑5, dem Nachfolger der Klassifikation DSM‑IV, ist die SAD als Unterkategorie der rezidivierenden Major Depression verzeichnet. Mit dieser Beschreibung wurde eine frühere Einschätzung revidiert, denn die Vorgängerin von DSM‑IV listete die SAD noch als eigenständige affektive Erkrankung auf.
Lichtmangel als Hauptstressor einer saisonal abhängigen Depression
Da der Lichtmangel jedoch das auffälligste Anzeichen des jahreszeitlichen Wechsels in nordamerikanisch-europäischen Breiten ist und auch mit nachgewiesenen Stoffwechselveränderungen einhergeht, wird er als SAD-Hauptstressor betrachtet. Lichtmangel ist ‑ neben jahreszeitlich bedingten Gründen ‑ auch die Folge eines zu häufigen Aufenthalts in geschlossenen Räumen und kann daher auch außerhalb der lichtarmen Jahreszeiten ein Problem darstellen. So betrifft Lichtmangel auch bettlägerige Personen oder Heimbewohner und überhaupt alle Personen, die sich zu wenig im Freien aufhalten. Es kann darüber hinaus ein relativer Lichtmangel bei Personen bestehen, die an das Leben in sonnenstarken Breitengraden angepasst sind und nun lichtärmere Gegenden bewohnen. Daher werden hier ausschließlich mögliche Folgen eines Lichtmangels für die psychische Gesundheit erörtert.
Erstmals erwähnte der griechische Arzt Hippokrates von Kos um 400 vor Chr. die Möglichkeit lichtmangelverursachter Affektstörungen, und wahrscheinlich behandelten die Ärzte der Antike depressive Patienten mit Hilfe des Sonnenlichts. Der im ersten oder zweiten Jahrhundert lebende griechische Arzt Aretaeus von Kappadokien, ein Anhänger von Hippokrates, befürwortete eine Therapie mit Licht.
Statistische Aussagen über die Verbreitung der saisonal abhängige Depression (SAD)
Wie viele Menschen in Deutschland von einer Lichtmangeldepression/SAD betroffen sind, ist unklar. So nennen die Stiftung Deutsche Depressionshilfe und auch das Informationsportal dasgehirn.info nur ca. ein Prozent der Allgemeinbevölkerung, was bei rund 82 Millionen Einwohnern immerhin noch etwa 820.000 Betroffene wären (Quellen: www.deutsche-depressionshilfe.de/stiftung/9732.php bzw. Christian Wolf, Licht im dunklen Winterloch, www.dasgehirn.info/...-5671). In populärwissenschaftlichen Beiträgen werden oft höhere Zahlen genannt, so berichtet Die Welt in einem Beitrag über etwa vier Millionen Menschen (Quelle: J. Zittlau, Winter macht dumm, müde und depressiv, 2013, WeltN24 GmbH, www.welt.de/.../article122719189...) und Bild der Wissenschaft nennt sogar die Zahl von 6% der Einwohner Deutschlands mit einer SAD (Quelle: C. Eberhard‑Metzger, Glück ist, wenn die Chemie stimmt, in: Bild der Wissenschaft, Konradin Medien GmbH, Leinfelden-Echterdingen 1999, www.wissenschaft.de/.../65655/).
Es scheint aber, dass in den Beiträgen nicht exakt zwischen Depression und depressiven Verstimmungen unterschieden wird, was die stark voneinander abweichenden Schätzungen erklären könnte.
Symptomorientierte Abrenzung der Lichtmangeldepression/SAD von der asaisonalen Depression
Mittels Symptomvergleichs erfolgt die Abgrenzung der durch Lichtmangel verursachten Affektstörung von einer asaisonalen Erkrankung. Beide Formen unterscheiden sich hinsichtlich Schlaf- und Appetitstörungen erheblich. Während bei Patienten mit einer asaisonalen Affektstörung verschiedene Formen von Schlaflosigkeit verbreitet sind (→ Abschnitt 1.1), tritt bei der Lichtmangeldepression ein übermäßig hohes Schlafbedürfnis auf, dem Betroffene auch folgen können. Allerdings scheint ihr Schlaf eher flach, so dass sie auch während des Tages müde und unausgeschlafen wirken. Der Appetit ist im Gegensatz zur asaisonalen Depression erhöht, betrifft besonders Kohlenhydrate und vor allem Süßigkeiten, was häufig auch mit einer erheblichen Gewichtszunahme verbunden ist.
Nach der ICD-Systematik können sämtliche Symptome einer Affekterkrankung bis zu einer mittelgradigen Schwere auftreten.
Empirische Untersuchungen allgemeiner Zusammenhänge zwischen Lichtmangel und Depression
Zunächst ein Überblick über einige wissenschaftliche Versuche, potentielle Zusammenhänge zwischen Lichtmangel und der Erkrankung an einer Affektstörung nachzuweisen. Wegen der Schwierigkeiten, jahreszeitbedingte oder breitengradabhängige (= latitudenabhängige) Lichtveränderungen getrennt von anderen jahreszeitlichen und latitudenbedingten Phänomenen zu untersuchen, werden vor allem Studien berücksichtigt, die sich mit der Wirkung des Lichts auf die Stimmungslage bei therapeutischer Anwendung beschäftigen.
Seit den ersten Überlegungen der beiden Ärzte der Antike, Hippokrates und Aretaeus, wurde der Gedanke über die Auswirkungen des Lichts auf den psychischen Gesundheitszustand über lange Zeit nicht bedeutend weiterentwickelt. Die Veröffentlichungen in der Neuzeit fanden erst zu Beginn des 19. Jahrhunderts statt, jedoch handeln sie von den potentiellen Auswirkungen des Jahreszeitenwechsels allgemein und nennen nicht den Lichtmangel explizit. So beschrieb der französische Psychiater Philippe Pinel als erster Fälle jahreszeitlich auftretender Affektstörungen anhand von Fallbeispielen. In Deutschland erwähnte Wilhelm Griesinger, ein Psychiater aus Württemberg, Beobachtungen über mögliche jahreszeitliche Zusammenhänge bei affektiven Erkrankungen in seinem im Jahre 1845 erschienenen Hauptwerk „Die Pathologie und Therapie der psychischen Krankheiten“ und bezieht sich dort insbesondere auf Jean‑Étienne Esquirol, einen Schüler Philippe Pinels (Quelle: W. Griesinger, Die Pathologie und Therapie der psychischen Krankheiten, 1845, Verlag Krabbe, Stuttgart).
Eine ernstzunehmende wissenschaftliche Auseinandersetzung mit dem Thema begann in jüngerer Zeit durch die Veröffentlichung Alfred J. Lewys im Jahre 1980. Lewy und sein Team, zu dem auch der noch zu nennende Thomas A. Wehr gehörte, bemerkten eine signifikante Reduzierung des Melatoninspiegels bei einer Exposition mit Licht von mehr als 2.500 Lux (Quelle: A. J. Lewy et al., Light suppress melatonin secretion in humans, 1980, Science 12/1980, S. 1.267/9, www.ncbi.nlm.nih.gov/pubmed...). Von dieser Studie erfuhr ein Jahr später Herbert Kern, der an sich selber eine regelmäßig im Winter auftretende Depression bemerkt hatte und einen Zusammenhang vermutete. Kern wandte sich an Dr. Lewy, worauf dieser ihn mit Licht behandelte. Das Ergebnis war eine angeblich komplette Remission der Depression Kerns. Lewy kreierte ‑ vielleicht etwas voreilig ‑ sogleich den Fachbegriff der „Seasonal Affective Disorder/SAD“ und publizierte den Fall im Jahre 1982. Die Lichttherapie war geboren.
Aufgrund der Arbeiten von A. J. Lewy wurde 1984 die Lichttherapie erstmals ausführlich von Forschern des US-amerikanischen National Institute of Menthal Health unter Führung des südafrikanischen Psychiaters Norman E. Rosenthal beschrieben, zu denen auch Alfred J. Lewy und Thomas Wehr gehörten (Quellen: The History of Moderne Biomedicine, www.histmodbiomed.org/.../vol51 und N. E. Rosenthal, J. C. Lewy, T. A. Wehr et al., Seasonal affective disorder: A description of the syndrome and preliminary findings with light therapy, 1984, Annals of General Psychiatry, 1/1984, S. 72 ‑ 80, www.ncbi.nlm.nih.gov/pubmed/6581756). Rosenthal fand 29 geeignete Patienten mittels Zeitungsanzeigen, deren meist Bipolare Affektstörungen in einem Zusammenhang mit saisonalen Schwankungen bzw. der geographischen Breite zu stehen schienen und die typische SAD-Symptome aufwiesen: hohes Schlafbedürfnis, Tagesschläfrigkeit und einen Hang zur übermäßigen und kohlenhydratreichen Ernährung. Bei 11 Patienten konnte ein depressionsmindernder Effekt durch eine Therapie mit Licht nachgewiesen werden.
Im Jahre 1987 wies eine Gruppe um den US-amerikanischen Psychiater Thomas A. Wehr nach, dass eine effektive therapeutische Wirkung des Lichts nur bei einer Aufnahme über die Netzhaut der Augen gewährleistet ist. So ging man bis dahin davon aus, dass die Effekte ausschließlich oder zum größten Teil auf die Sonnenbestrahlung der Haut zurückzuführen sind (Quellen: Siegfried Kasper, Jahreszeit und Befindlichkeit in der Allgemeinbevölkerung, 1991, Springer‑Verlag Berlin/Heidelberg und Thomas A. Wehr, Norman E. Rosenthal et al., Eye versus skin phototherapy of seasonal affective disorder, The American Journal of Psychiatry 1987, 144, S. 753 ‑ 757, www.ncbi.nlm.nih.gov/pubmed/3591996).
Die Wirkungen des Lichts bei affektiv Erkrankten wurden seit den ersten Untersuchung von Rosenthal und Wehr häufig erforscht. Eine gute Übersicht über verschiedene Lichttherapiestudien ermöglicht die Online-Publikation der BMJ Publishing Group Ltd mit einem Beitrag des industrieunabhängigen Drug and Therapeutics Bulletin, in der auch die beiden nachfolgenden Quellen aufgeführt sind (Quelle: Management of seasonal affective disorder, 2010, BMJ, www.bmj.com/content/340/bmj.c2135).
Eine systematische Überprüfung acht randomisierter und kontrollierter Studien mit insgesamt 360 Personen und Lichtherapien zwischen 7 und 42 Tagen mit mindestens 3.000 Lux Lichtstärke lässt auf eine Wirksamkeit schließen. In fünf Studien mit 133 Personen konnte auch die Wirksamkeit einer Dämmerungssimulation nachgewiesen werden, bei der man die Lichtstärke innerhalb von 1 1/2 bis 2 1/2 Stunden von 0 auf 200 bis 300 Lux ansteigen ließ. Dadurch konnten die SAD-Symptome mittel bis stark („moderate to large“) reduziert werden (Quelle: Robert N. Golden et al., The efficacy of light therapy in the treatment of mood disorders: A review an meta-analysis of the evidence, 2005, 162/4, S. 656 ff., American Journal of Psychiatry, www.ncbi.nlm.gov/pubmed/...).
Keine klare Favorisierung ergab eine 20 Einzelstudien umfassende Meta-Studie, bei denen die Wirksamkeit einer Anwendung von Licht und weiteren Therapieformen (Verhaltenstherapie, Therapie mit Antidepressiva) bzw. Placebo-Behandlungen mit funktionslosen Lichtboxen und den unbehandelten Patienten auf einer Warteliste untersucht wurden. Alle Formen, einschließlich der Placebo-Therapie, erwiesen sich als gleich wirksam. Studien über direkte Vergleiche zwischen einer Lichttherapie und alternativen Behandlungsformen wurden noch nicht durchgeführt (Quelle: National Institute for Health and Clinical Excellence, Depression in adults - update 2009).
Ein Forscherteam der US-amerikanischen University of Alabama unter Leitung von Shia T. Kent veröffentlichte im Jahre 2009 im Online-Portal Environmental Health die Ergebnisse ihrer Studie über die kognitiven Auswirkungen des Sonnenlichts auf Menschen mit und ohne diagnostizierter Depression, wobei nicht zwischen saisonaler und asaisonaler Depression unterschieden wurde. Kent ging von einem kausalen Zusammenhang zwischen der Menge an Sonnenlicht und der Serotonin- bzw. Melatoninregulation aus. Serotonin und Melatonin sollen auch bei kognitiven Funktionen eine Rolle spielen (Quelle: Shia T. Kent et al., Effect of sunlight exposure on cognitive function among depressed and non-depressed participants: a REGARDS cross-sectional study, 2009, Environ Health, www.ncbi.nlm.nih.gov/pmc/articles/PMC2728098/).
Die Forscher untersuchten planerisches Denken und das Kurzzeitgedächtnis von 16.800 Patienten ab 45 Jahren in Abhängigkeit der Sonneneinstrahlung. Nach Ausschluss von 2.326 Teilnehmern verblieben in der Studie 14.474 Personen. Der Grad der Depressivität wurde auf Basis eines Fragenbogens und einer gängigen Bewertungsskala ermittelt. Auch für die Ermittlung der kognitiven Fähigkeiten wurde ein Fragebogen verwendet. Die Wetterdaten zur Sonnenexposition lieferte die NASA.
Im Ergebnis konnte ein Zusammenhang zwischen einer verringerten Sonnenstrahlung über zwei Wochen und dem Rückgang kognitiver Fähigkeiten bei depressiven Studienteilnehmern festgestellt werden. Interessanterweise waren von den kognitiven Einschränkungen auch Patienten mit einer asaisonalen Depression betroffen. Bei den nichtdepressiven Teilnehmern kam es zu keinerlei Einschänkungen.
In Norwegen, einem Land, deren Bewohner aufgrund der geographischen Lage immer mit sehr langen und dunklen Wintern konfrontiert werden, verneint der Mediziner Vidje Hansen die Existenz einer lichtmangelbedingten Depression: „Es gibt keine Winterdepression, glaubt der Psychiater Prof. Vidje Hansen von der Universitätsklinik Tromsø: «Das ist eine erfundene Krankheit. Als diese These 1984 in Amerika aufgestellt wurde, waren wir im Norden Norwegens sehr überrascht, denn wir Psychiater sehen nicht, dass die Menschen im Winter häufiger über Depressionen klagen als in anderen Jahreszeiten.» Die norwegische Stadt liegt zwei Monate im Jahr im anhaltenden Dunkel der Polarnacht. «Seit 1980 haben wir 30.000 Leute nach ihren Problemen in der Winterzeit befragt», erinnert sich Hansen. «Ein Fünftel leidet unter Schlafstörungen und dauernder Müdigkeit, aber für Depressionen fanden wir keinerlei Anzeichen.» Er glaubt, dass die früheren Studien einfach ungenau waren. «Wenn überhaupt, könnte die Stimmung vom Klima abhängen, nicht so sehr von der Dunkelheit.» 1998 kamen andere Forscher in Tromsø noch zu einem anderen Ergebnis: Im Raum Oslo litten 16,9 Prozent der Bewohner unter Winterdepressionen, jenseits des Polarkreises 21,2 Prozent, stellenweise bis zu 37 Prozent. Viele Menschen mögen schlicht bestimmte Witterungen nicht, meint Hansen: «Im Oktober und November - da ist das Wetter in Italien schlecht - erscheinen Italiener stärker winterdepressiv als die Menschen in Tromsø.» Rentiere haben sich im Laufe der Evolution an die Dunkelheit angepasst, weiß Prof. Karl‑Arne Stokkan vom Institut für arktische Biologie der Universität Tromsø: «Sie sind jeweils sechs bis acht Stunden aktiv und schlafen dann drei, vier oder sechs Stunden. Sie haben keine regelmäßigen Schlafphasen alle 24 Stunden wie wir Menschen.»“ (Quelle: „Die Winterdepression ist eine erfundene Krankheit“ , Online-Beitrag auf der Webseite der nano-Redaktion des Fernsehsenders 3sat, 1/2013).
Lichtmangel als kausaltheoretisch begründbare Noxe einer Affektiven Störung?
Einige der zitierten Studien lassen auf einen kausalen Zusammenhang zwischen Lichtmangel und affektiven Erkrankungen schließen. Ein solcher Zusammenhang lässt sich physiologisch nur begründen, wenn Lichtmangel nachweisbar direkte oder indirekte Wirkungen auf affektrelevante Hirnareale hat.
Folgende sechs Aspekte, die alle mit Licht bzw. der Lichtstärke in Zusammenhang stehen, sind zu diskutieren:
- Sinkender Vitamin-D3-Spiegel (Calciolmangel)
- Störungen der Serotoninregulation
- Melatonin und Melanopsin
- Evolutorisch bedingter Rückgang von Aktivitäten im Winter
- Genetische Zusammenhänge
- Reaktive Aspekte
Zu 1.: Sinkender Vitamin-D3-Spiegel (Calciolmangel)
Die Calciolbildung (Vitamin D3) erfolgt mit Hilfe des in Darmschleimhaut und Leber produzierten Ausgangsstoffes 7‑Dehydrocholesterol (Provitamin D), der sich in großen Mengen in der Basal- und Stachelzellschicht der Haut ansammelt und dort mit Hilfe der im Tageslicht enthaltenen UV‑B‑Strahlung (Wellenlänge zwischen 285 und 315 nm) zu Calciol umgewandelt wird. Um im Stoffwechsel aktiv werden zu können, muss Calciol in einem weiteren Schritt zu Calcitriol umgewandelt werden. Letzteres spielt hier jedoch keine Rolle, die Bezeichnung D3 wird der Einfachheit halber synonym für Calciol und Calcitriol verwendet.
Die Menge des in der Haut produzierten Vitamins D3 ist wichtig für den gesamten Organismus, denn nur ein ganz geringer D3‑Anteil von 10% kann über die Nahrung aufgenommen werden (Quelle: P. Knuschke et al., UV‑abhängige Vitamin D Synthese ‑ Bilanzierung der Expositionszeit durch UV zur Produktion des optimalen Vitamin D3‑Bedarfs im menschlichen Körper, Ressortforschungsbericht zur kerntechnischen Sicherheit und zum Strahlenschutz des Bundesamts für Strahlenschutz, 2007 ‑ 2011, Technische Universität Dresden und Medizinische Fakultät Carl Gustav Carus, Klinik und Poliklinik für Dermatologie, https://doris.bfs.de/...).
Es gibt es drei Ursachen für eine D3-Minderproduktion:
- Nicht ausreichende UV‑B‑Strahlung
Die häufigste Ursache einer gestörten Calciol-Synthese ist das Fehlen von Tages- bzw. Sonnenlicht, da bei ungenügender UV‑B‑Strahlung die Umwandlung in Calciol in der Haut sinkt. Dies konnte in Studien nachgewiesen werden, unter anderem in der oben zitierten Arbeit von P. Knuschke et al. des Bundesamts für Strahlenschutz. Während der lichtarmen Jahreszeiten sinkt der UV‑B‑Anteil des Tageslichts dramatisch ab, denn aufgrund des tiefen Sonnenstands dringt davon kaum noch etwas davon durch die Atmosphäre. In der Studie konnte gezeigt werden, dass der D3-Blutspiegel bei den Probanden im Winter im Schnitt mit 18 ng/ml unterhalb des Mindestwertes von 20 ng/ml lag.
Menschen, die sich häufig oder überwiegend in geschlossenen Räumen aufhalten, haben mit hoher Sicherheit eine ungenügende Calciolproduktion. Darüber hinaus wirkt Fensterglas wie ein UV‑B‑Filter; es nützt also auch nichts, sich dort in der Nähe von Fenstern aufzuhalten.
Aber auch im Sommer hat der Körper häufig keine Möglichkeit, Calciol zu produzieren, denn zum Schutz vor Sonnenstrahlung bedecken viele Menschen eine zu große Hautfläche mit Textilien. Der Restanteil der Haut, der noch dem Sonnenlicht ausgesetzt ist, wird dann mit Sonnenschutzmitteln gegen UV‑B‑Strahlung geschützt, die ab Lichtschutzfaktor 8 keine Strahlung mehr durchlassen. Häufig vermindern im Sommer Smog und Ozon die UV‑B‑Strahlung zusätzlich.
Das fettlösliche Vitamin D3 wird im Körper gespeichert. Das in der lichtintensiven Jahreszeit gespeicherte Vitamin dient als Puffer für die lichtarmen Perioden, allerdings nur dann, wenn eine ausreichende Menge gespeichert werden konnte.
Auch eine Gegensteuerung mit Hilfe von Solarien nützt nichts, denn die meisten von ihnen verwenden ausschließlich die bräunende UV‑A‑Strahlung ohne einen UV‑B‑Anteil. Und da in der gebräunten Haut noch weniger Calciol produziert wird, kann der Gang zum Sonnenstudio sogar kontraproduktiv sein.
- Dunkle Hautfarbe
Das leitet über zur Hautfarbe. Die Haut von Menschen, die in Breitengraden rund um den Äquator beheimatet sind, hat sich an das dort übliche starke Sonnenlicht angepasst ‑ je höher die UV‑Strahlung, desto dunkler die Hautfarbe. Dunkle Haut produziert bei gleicher Menge an UV‑B‑Strahlung weniger Calciol. Verlassen Menschen mit dunkler Hautfarbe ihre Heimat, um in äquatorfernen Ländern zu leben, vergrößert sich deren Risiko, einen Calciolmangel zu erleiden.
- Hauterkrankungen und die fortgeschrittene Alterung von Haut, Leber und Darm
Ältere Menschen produzieren mit ihrer gealterten Haut weniger Calciol, die Calciol-Syntheserate sinkt auf etwa 25%. Laut der Studie des Bundesamtes für Strahlenschutz kann das jedoch nicht an der Einlagerung von zu wenig 7‑Dehydrocholesterol liegen, denn diese zeigte keine Altersabhängigkeit, so dass hier andere Ursachen infrage kommen.
Eine großflächige Hauterkrankung kann einen vergleichbaren Effekt haben, denn sie behindert den Calciol-Syntheseprozess direkt und/oder die Einlagerung der Ausgangssubstanz 7‑Dehydrocholesterol in der Haut.
Eine ungenügende Einlagerung des Ausgangsstoffes 7‑Dehydrocholesterol ist auch bei einer Erkrankung oder aufgrund der fortgeschrittenen Alterung von Leber oder Darm(‑schleimhaut) möglich, da eventuell zu wenig der Ausgangssubstanz produziert wird.
Die Notwendigkeit der im Tageslicht enthaltenen UV‑B‑Strahlung für die Produktion von genügend D3 ist demnach unzweifelhaft. Aber kann ein D3‑Mangel auch Affektstörungen begünstigen?
Die Funktionen der zu Calcitriol umgewandelten bioaktiven Substanz und Zusammenhänge mit affektiven Erkrankungen werden in den Abschnitten 2.3.1 und 4.4 thematisiert, die wichtigsten Fakten sind nachfolgend zusammengefasst.
D3 ist am Hirnzellenwachstum (Nerven- und Gliazellen) und an der Reizweiterleitung beteiligt. Der Nachweis der Beteiligung an der Reizleitgeschwindigkeit wurde bisher nur bei motorischen Nervenzellen erbracht, jedoch ist eine solche Funktion auch für andere Neuronentypen denkbar.
In affektrelevanten Gehirnarealen befinden sich Rezeptoren für Calcitriol, beispielsweise im Thalamus und Hippocampus. Auch die Neurotransmitter-Synthese könnte D3 laut einiger Studien mitverantworten. Dass D3 auch bei der Transkription in Nerven- bzw. Gliazellen benötigt wird, ist (noch) nicht bestätigt, jedoch gibt es Nachweise für andere Organe.
Potentielle D3‑Mangelerscheinungen im Zentralnervensystem sind
- neben der Depression - Denkstörungen, Krampfanfälle, Muskelkrämpfe, niedriger Muskeltonus, Psychosen und Schlafstörungen, letztere jedoch eher in der Form der Schlaflosigkeit, so wie sie von asaisonalen Affektstörungen bekannt ist.
In der empirischen Forschung bemüht man sich, Zusammenhänge zwischen einem Vitamin‑D3‑Mangel und Affektstörungen zu untersuchen und ein solcher wird immer wieder festgestellt. Hier beispielhaft eine Übersicht über zwei Meta-Studien:
Eine Meta-Studie am New Yorker Columbia University Medical Center (Quelle: J. A. Shaffer, D. Edmondson et al. 2013: Low vitamin D and depression, A systematic review and meta-analysis, Center for Behavioral Cardiovascular Health, Columbia University) untersuchte drei Veröffentlichungen (Chan et al. 2012, May et al. 2010 und Milaneschi et al. 2010), die an Personen mit einem durchschnittlichen Alter von mehr als 70 Jahren durchgeführt wurden. Sie kamen zu dem Ergebnis, dass bei diesen Individuen mit einem Vitamin‑D‑Defizit ein 2,3 fach höheres Risiko besteht, an einer Depression zu erkranken und konstatierten einen Bedarf für weitere Untersuchungen. Leider kann wegen der Bedingungen der zugrundeliegenden Studien nicht auf die Situation jüngerer Personen unter 50 Jahren geschlossen werden und auch kausale Zusammenhänge erschließen sich durch derartige Übersichtsstudien nicht.
Im selben Jahr kamen andere Forscher zu einem ähnlichen Ergebnis, wobei ihre umfangreichere Meta-Studie 14 Untersuchungen unterschiedlichen Studien-Designs mit insgesamt über 31.000 Teilnehmern zugrunde legte und auch jüngere Menschen umfasste. Es wurden geringere Vitamin‑D‑Spiegel bei depressiv erkrankten Patienten gefunden als bei gesunden Personen und ebenfalls ein erhöhtes Quotenverhältnis bzw. eine erhöhte Hazard Ratio bei Depression von niedrigen zu höchsten Vitamin‑D‑Spiegeln festgestellt. Ein niedriger Spiegel steht demnach mit Depressionserkrankungen in einem signifikanten Zusammenhang. Eine Aussage bezüglich der Kausalität dieser Assoziation ist nach den Schlussfolgerungen der Autoren auf Grundlage weiterer Studien zu klären, bei denen Therapien mit Vitamin D untersucht werden müssten (Quelle: Rebecca Anglin, Zainab Samaan et al. 2013, Vitamin D deficiency and depression in adults: Systematic review and meta-analysis, The British Journal of Psychiatry 2013, 202, S. 100 - 107, doi: 10.1192/bjp.bp.111.106666).
Quintessenz: Die Ergebnisse der Forschungsarbeiten über die Abhängigkeit der D3-Produktion von der Sonnenstrahlung zeigen, dass Lichtmangel zu D3-Fehlversorgungen führen kann. Aufgrund der Aufgaben des Calciols bzw. des bioaktiven Calcitriols im Zentralnervenssystem dürfte auch dort mit entsprechenden negativen Auswirkungen zu rechnen sein.
Beispielhaft sei hier auf den Hippocampus und den Thalamus mit ihren Calcitriol-Rezeptoren verwiesen. Der Hippocampus ist u. a. sowohl für die Verarbeitung von Emotionen und Affekten als auch das Gedächtnis wichtig, der Thalamus mit seiner Filterfunktion für das zielgerichtete Handeln. Wie die oben zitierte Studie der University of Alabama unter Leitung von Shia T. Kent zeigt, scheinen auch diese Funktionen lichtabhängig zu sein.
Es ist daher plausibel, dass Lichtmangel über die D3-Mangelversorgung die Entstehung einer Affektstörung (mit‑)verantwortet bzw. begünstigt oder die Verstärkung der Symptome einer schon bestehenden Erkrankung bewirkt.
Zu 2.: Störungen der Serotoninregulation
Synthese und Funktionen des Neurotransmitters Serotonin werden in Kapitel 5 detaillierter beschrieben. Zum Verständnis dieses Abschnitts soll hier die Darstellung der wichtigsten Fakten genügen.
Die Serotoninproduktion basiert auf der Aminosäure L‑Tryptophan. Die größte Menge ‑ ca. 90 bis 95% ‑ produziert und speichert der Verdauungstrakt. Sie wird für die Regulation verschiedener Körperfunktionen genutzt, beispielsweise die Darmperistaltik, den Blutdruck durch die Gefäßeng- oder weitstellung und die Blutgerinnung. Dieses Körper-Serotonin kann die Blut‑Hirn‑Schranke nicht überwinden und hat daher im Zentralnervensystem keine Bedeutung.
Nur etwa 5 bis 10% des gesamten Serotonins werden im Gehirn in den serotonergen Nervenzellen des gesamten Stammhirns produziert, die sich in den Raphe-Kernen befinden und über Nervenbahnen mit vielen anderen Teilen des Gehirns verbunden sind. Es existiert ein Regelkreis, der diese Produktion überwacht, wobei Serotonin-Autorezeptoren im Stammhirn dabei eine zentrale Funktion ausüben, indem sie bei einer Aktivierung durch Serotonin dessen Produktion drosseln.
Die zellphysiologische Aufgabe des Serotonins im Gehirn ist die Reizübertragung zwischen zwei Nervenzellen. Serotoninmoleküle in den Endknöpfchen der Überträger- bzw. Senderzelle werden in einen synaptischen Spalt abgegeben und bewegen sich dann in Richtung der Dendriten der Empfängerzelle. Dort befinden sich serotoninspezifische Kontaktstellen (Rezeptoren), an denen die Serotoninmoleküle andocken und in der Empfängerzelle physiologische Prozesse auslösen. Die Serotoninmoleküle werden anschließend durch Transportproteine über spezielle Rezeptoren an den Endknöpfchen der Überträgerzellen wieder in diese zurückgeführt und dort größtenteils für eine erneute Ausschüttung in Vesikel eingelagert oder mit Hilfe des Enzyms Monoaminoxydase (MAO) abgebaut.
Es ist kein unmittelbarer Zusammenhang zwischen der Serotoninproduktion und einer Lichteinwirkung nachgewiesen, so wie es von der Calciol-Synthese (Vitamin D3) in der Haut bekannt ist, denn dafür ist der UV‑B‑Anteil des Tageslichts zwingend erforderlich. Es gibt jedoch Anhaltspunkte dafür, dass Licht einen indirekten Einfluss auf den Serotoninstoffwechsel hat.
Im Jahre 2008 stellten Jeffrey H. Meyer und Wissenschaftler der Universitäten Toronto und Wien an 88 Probanden fest, dass präsynaptisch die Bindungspotentiale der Serotonin-Transportproteine im Winter wesentlich höher waren als im Frühjahr und Sommer (Quelle: J. H. Meyer, S. Houle, A. A. Wilson et al., Seasonal Variation in Human Brain Serotonin Transporter Binding, Archives of General Psychiatry, 9/2008, 65, S. 1072 ‑ 78, Chicago/USA 2008). Es könnten dadurch im Winter vermehrt Serotoninmoleküle zurück in die Senderzellen transportiert werden, so dass die Serotoninmenge im synaptischen Spalt reduziert ist, womit sich eine saisonale Abhängigkeit des Serotoninstoffwechsels begründen ließe. Die Studie wurde jedoch ausschließlich mit gesunden Teilnehmern erstellt und die exakten physiologischen Gründe für das erhöhte Bindungspotential des Transportproteins im Winter waren auch nicht Inhalte der Untersuchung. Ob die Serotoninkonzentration im synaptischen Spalt bei einer erhöhten Transportaktivität tatsächlich geringer ist, ist spekulativ. Denn die Frage, ob sich parallel dazu mittels des Serotoninregelkreises auch die Serotoninmenge erhöht, bleibt unbeantwortet. Insgesamt können Schlussfolgerungen hinsichtlich der Wirkungen eines Lichtmangels auf den Serotoninstoffwechsel aufgrund dieser Studie daher nur vage sein.
Mit einer Studie aus dem Jahre 2011 haben der Psychiater C. J. Spindelegger und ein Team der Medizinischen Universität Wien auch postsynaptische Veränderungen an den Zielzellen des Serotoninübertragungsprozesses nachgewiesen, die auf Lichtmangel beruhen sollen. 1A‑Serotoninrezeptoren postsynaptischer Neuronen hatten in den emotionsverarbeitenden Bereichen bei Lichtmangel ihr Bindungspotential verringert (Quelle: C. Spindelegger, P. Stein, S. Kaper et al., Light-dependent, alteration of serotonin-1A receptor binding in cortical and subcortical limbic regions in the human brain, The World Journal of Biological Psychiatry, 9/2012 13 (6), S. 413 ‑ 22, London/UK 2012). Die Studie wurde ebenfalls ausschließlich an 36 gesunden Probanden durchgeführt. Auch hier waren die genauen Mechanismen der physiologischen Änderungen nicht Untersuchungsgegenstand, ebensowenig etwaige Kompensationsmechanismen aufgrund des unterstellten Effekts.
Ein weiterer potentieller Grund für einen geringen Serotoninspiegel bei Lichtmangel könnte ein Verdrängungseffekt durch die gesteigerte Melatoninproduktion sein. Das Hormon Melatonin wird bei Dunkelheit hauptsächlich in der Epiphyse auf der Basis von Serotonin synthetisiert (→ nachfolgenden Abschnitt „Melatonin und Melanopsin“). Daraus könnte man folgern: Die Melatonin-Synthese geht unmittelbar auf Kosten der Serotoninproduktion.
Theoretisch kommt es jedoch nur zu einem Serotoninmangel, wenn die Regulierung der Serotoninproduktion in den Raphe-Kernen des Mittelhirns oder der Medulla oblongata nicht adäquat funktioniert und nicht auf den sinkenden Serotoninspiegel reagiert. Hierfür gibt es jedoch keine Belege. Dem Autoren sind keine Studien bekannt, die sich mit dieser Frage beschäftigen. Desweiteren könnte die Verdrängung von Serotonin durch Melatonin auch gewünscht sein und damit einen Teil des Serotonin-Regelkreises repräsentieren.
Vermutungen über Zusammenhänge zwischen der Lichteinwirkung und der Serotoninsynthese sind damit äußerst vage.
Die zweite - und noch wichtigere Frage - ist aber, ob Serotonin bei der Affekt- und Angstverarbeitung überhaupt eine Rolle spielt.
Im ersten Kapitel wurde die kontroverse wissenschaftliche Diskussion um Serotonin schon erwähnt, sowohl hinsichtlich der Monoaminmangelhypothese als auch der Wirksamkeit von Medikamenten, die den Serotoninstoffwechsel manipulieren (→ Abschnitte 1.1 f.und 1.2).
Die Meinungen gehen weit auseinander. Ein Artikel des Magazins Zeit Wissen aus dem Jahre 2008 fasst die Auseinandersetzung sehr anschaulich in wenigen Worten zusammen und ist immer noch aktuell: „Der Botenstoff Serotonin hat eine erstaunliche Karriere als Glückshormon hingelegt. Maßgeblich beteiligt daran waren Werbemaßnahmen für Medikamente. Seit 1965 verdächtigen Ärzte einen niedrigen Serotonin-Spiegel im Gehirn, für Depressionen verantwortlich zu sein. Doch die große Zeit des Botenstoffs begann in den 80er Jahren. Damals wurde eine neue Art von Antidepressiva entwickelt, die Selektiven Serotonin-Wiederaufnahme-Hemmer (SSRI, berühmt wurde vor allem Prozac, in Deutschland unter dem Namen Fluctin im Handel). Diese sollen verhindern, dass die Nervenzellen im Gehirn das ausgeschüttete Serotonin zu schnell wieder aufnehmen. Auf der deutschen Internetseite des Pharmaherstellers Pfizer steht nach wie vor: «Bei einer Depression besteht ein Mangel an Serotonin.» Doch dies konnte bis heute keine Studie nachweisen. Manche Depressive haben sogar einen höheren Serotonin-Spiegel als Gesunde. Was ein normaler Wert ist, weiß ohnehin niemand. Außerdem dauert es in der Regel mehrere Wochen, bis die SSRI bei Patienten Wirkung zeigen, obwohl die Wiederaufnahme-Hemmung schon nach den ersten Tabletten einsetzt. Viele Forscher vermuten inzwischen, dass Serotonin nur indirekt mit der Depression zu tun hat. Die irische Medikamenten-Aufsichtsbehörde jedenfalls untersagte 2003 einem Pharmahersteller, in Informationsbroschüren zu behaupten, das Antidepressivum korrigiere ein chemisches Ungleichgeweicht im Gehirn.“ (Quelle: Eva‑Maria Schnurr, Gefährliche Helfer, Zeit Wissen 2/2008, Zeitverlag Gerd Bucerius, Hamburg).
Nach wie vor ist es also lediglich eine Vermutung, dass Serotonin im Zusammenhang mit Affekten und Ängsten eine bedeutende Rolle spielt. Erst recht gilt das für kausale Zusammenhänge zwischen Serotonin und Affektstörungen, die nach wie vor nicht belegt sind. Auch die Überprüfungen der Wirksamkeit von SSRI in verschiedenen Studien deuten auf keinen bzw. einen nur schwach ausgeprägten Zusammenhang hin (→ Abschnitt 1.1). In ihrem Artikel erwähnt die Autorin auch, dass viele Wissenschaftler eher von einem indirekten bzw. korrelativen Zusammenhang ausgehen. Nach deren Ansicht würden Veränderungen des Serotoninstoffwechsels und eine Depression lediglich auf gleiche Ursachen zurückzuführen sein.
Quintessenz: Ein Zusammenhang zwischen Lichtmangel und Affektstörungen aufgrund lichtbedingter Veränderungen im Serotoninstoffwechsel bleibt strittig. Eine direkte Einflussnahme des Lichts auf die Serotonin-Synthese findet nicht statt. Die Studienlage über Nachweise indirekter Einflüsse des Lichts durch prä- und postsynaptische Veränderungen des Serotoninstoffwechsels sind noch zu dürftig. Über einen unerwünschten Verdrängungseffekt der Serotoninproduktion durch Melatonin ist nichts bekannt. Und die grundsätzliche Frage der Funktionen des Serotonins bei der Verarbeitung von Emotionen und Ängsten ist noch gänzlich unbeantwortet.
Zu 3.: Melatonin und Melanopsin
Sind also Serotoninverdrängungseffekte aufgrund einer potentiell konkurrierenden Melatoninproduktion als Ursache einer Depression eher unwahrscheinlich, könnte es dagegen einen direkten Zusammenhang zwischen der lichtabhängigen Melatoninproduktion und Affektstörungen geben. In einer der oben erörterten Studien wurde darauf schon Bezug genommen (Quelle: A. J. Lewy et al., Light suppress melatonin secretion in humans, 1980, Science 12/1980, S. 1.267/9, www.ncbi.nlm.nih.gov/pubmed...).
Die Melatonin-Synthese setzt bei beginnender Abenddämmerung bzw. nachts oder bei Dunkelheit in der Epiphyse ein, die dann das Hormon ohne Speicherung verstärkt in den Blutkreislauf sezerniert. Der Melatoninspiegel steigt dabei auf das 10fache des Tageswerts an.
Der Regelprozess wird von speziellen lichtempfindlichen Ganglienzellen auf der Augennetzhaut angestoßen. Bei Tageslicht produzieren sie Melanopsin und senden damit ihre Lichtinformationen über den Sehnerv an den Hypothalamus. Der stoppt daraufhin die Melatoninproduktion. Bei entsprechend wenig Tageslicht wird die Melatoninproduktion in der Epiphyse hingegen angeregt.
Leider sind die Aufgaben von Melatonin nicht vollständig bekannt. Mit ziemlicher Sicherheit kann man davon ausgehen, dass Melatonin dem Körper den Beginn von Dämmerung und Dunkelheit signalisiert und an der Umstellung verschiedener Körperfunktionen auf Nachtbetrieb ‑ auch zirkadianer Rhythmus genannt ‑ beteiligt ist: Herzschlagfrequenz, Blutdruck, Verdauungsaktivitäten und Körpertemperatur sinken.
Die weit verbreitete Annahme, Melatonin mache müde, ist unter Fachleuten umstritten. So wird die Bezeichnung von Melatonin als Schlafhormon beispielsweise vom Berliner Schlafforscher Dieter Kunz abgelehnt (Quelle: Lydia Klöckner, „Wir befinden uns im Schlafmodus“, Zeit Online vom 5.11.2012).
Aber selbst bei der Annahme, das Hormon mache schläfrig, ist damit noch nicht dessen direkte oder indirekte Beteiligung an einer Lichtmangeldepression nachgewiesen. Ein solcher Zusammenhang kann nur unter entsprechenden Studienbedingungen geprüft werden.
Für die häufiger gemachte Vermutung, Melatonin senke den Antrieb, gibt keine Beweise.
Alle Studien, die einen Nachweis des Zusammenhangs zwischen einer Lichttherapie und der Verbesserung der depressiven Symptomatik führen, helfen hier nicht weiter. Denn: Veränderungen der Melatoninmenge sind nur eine mehrerer Folgen der Lichtveränderung im Organismus. Es ist daher spekulativ, zu behaupten, die Verschlechterung der Stimmungslage wäre eine unmittelbare Folge der Melatoninausschüttung.
Es sind nur Studien aussagekräftig, die einen direkten Zusammenhang zwischen der Einnahme von Melatonin und Veränderungen der affektiven Symptomatik untersuchen. Die beiden folgenden Mini-Studien haben einen solchen untersucht, sind jedoch aufgrund des unzureichenden Studiendesigns nur sehr gering beweiskräftig. Interessant ist jedoch, dass sie - wenn überhaupt - einen umgekehrten Zusammenhang feststellten, denn die Depressionssymptomatik verringerte sich durch die Einnahme von Melatonin.
Im Jahre 1997 fanden Alfred J. Lewy und sein Team von der Oregon Health and Sciences University heraus, dass niedrige Melatoningaben die Symptome von SAD-Patienten reduziert. Leider umfasste die Pilotstudie nur zehn Personen, davon erhielten fünf Melatonin und fünf ein Placebo. Lewy weist darauf hin, dass die Ergebnisse mit einer umfangreicheren Anlayse überprüft werden sollten (Quelle: A. J. Lewy, V. K. Bauer, N. L Cutler, R. L. Sack, Melatonin treatment of winter depression: a pilot study, Sleep and Mood Disorders Laboratory, Dep. of Psychiatry, Oregon Health and Sciences University, Portland/Oregon, USA, 10/1997, https://www.gwern.net/.../1998-lewy.pdf).
E. J. Dalton et al. führten im Jahre 2000 eine vierwöchige offene Studie mit acht behandlungsresistenten depressiven Patienten durch. Diese erhielten zusätzlich zu ihrer Standardmedikation 5 bis 10 mg Melatonin. Die Bewertung der Schwere der Depression erfolgte mit Hilfe der Hamilton-Depressionsskala (HAMD) vor und nach der Behandlung. Im Durchschnitt verringerte sich der HAMD-Index um 20%, wobei die Streuung zwischen 0 und 45% lag, also bei keinem 50% oder mehr erreicht wurden. Darüber hinaus stellte man einen leichten Rückgang der Schlafprobleme fest. Aufgrund des mageren Studien-Designs sind die Ergebnisse auch dieser Studie mit Vorsicht zu behandeln (Quelle: Dalton, Rotondi, Levitan, Kennedy, Brown, Use of slow-release melatonin in treatment-resistant depression, Depression Clinic, Centre for Addiction and Mental Health Toronto, Journal of Psychiatry Neuroscience, 1/2000, 25 (1), S. 48 - 52, http://www.ncbi.nlm.nih.gov/...PMC1407707/).
Melanopsin spielt in den bisher erörterten Zusammenhängen lediglich eine Rolle als Lichtinformationsprotein der Augennetzhaut. Könnte es auch noch andere Aufgaben bei der Regulierung des Tag-Nacht-Rhythmus haben?
Eine in Finnland durchgeführte Untersuchung von neun Gehirnen Verstorbener ergab, dass Melanopsin in vielen Regionen zu finden ist. Dies könnte auf eine generelle Lichtempfindlichkeit des Gehirns hindeuten. Welche Auswirkungen das auf den zirkadianen Rhythmus oder Affekte hat, ist nicht geklärt (Quelle: Nissilä, Mänttäri, Tuominen et al., The abundance and distribution of melanopsin (OPN4) protein in human brain, University of Oulu/Finland, European Psychiatry, Vol. 27, Supplement 1, 2012, http://www.sciencedirect.com/...).
Melanopsin könnte im Zentralnervensystem auch als gewöhnlicher Botenstoff wirken, dessen Funktionen aber auch ungeklärt sind.
Quintessenz: Einen kausalen Zusammenhang zwischen einer Depression und der Erhöhung des Melatoninspiegels könnte es aufgrund der Funktionen des Hormons geben, er wurde jedoch noch nicht zweifelsfrei bestätigt. In kleineren Studien kam es nach Melatoningaben zu einem umgekehrten Effekt. Diese Untersuchungen sind aufgrund der geringen Teilnehmeranzahl jedoch wenig aussagekräftig. Melanopsin wurde im Gehirn nachgewiesen, dessen Funktion sind jedoch unklar.
Zu 4.: Evolutorisch bedingter Rückgang von Aktivitäten in lichtarmen Jahreszeiten
Da die Aktivitäten vieler Tiere und Pflanzen in der lichtarmen Zeit im Winter generell sinken, teilweise bis zum extremen Rückzug in den Winterschlaf oder durch den Verlust des Blattwerks, könnte man auf vergleichbare Auswirkungen auch beim Menschen schließen.
Und gerade deswegen drängt sich der Gedanke auf, während der dunklen Jahreszeiten könnte eine Erhöhung des Depressionsrisikos steigen. Denn während beispielsweise Tiere dem Rückgang ihres Antriebs instinktiv folgen, funktioniert das bei Menschen in den westlichen und westlich orientierten Gesellschaften nicht oder nur bedingt. Menschen müssen hier auch im Winter auf Hochtouren laufen, obwohl ihr Organismus seine Aktivitäten eigentlich herunterfahren möchte.
Einige Wissenschaftlicher vermuten, dass sich diese ständige Auflehnung gegen die vom Körper geforderte Ruhe während der lichtarmen Zeit über Jahre zu einem massiven psychosozialen Stressfaktor entwickeln könnte, der die Entstehung einer saisonalen Affekterkrankung begünstigt, die langfristig auch in einer asaisonalen Affekterkrankung enden kann. Dem Autoren sind jedoch keine Studien o. ä. bekannt, die einen solchen Zusammenhang belegen.
Quintessenz: Dass es einen evolutorisch begründeten Zwang für Menschen gibt, in den lichtarmeren Jahreszeiten ihre Aktivitäten zurückzufahren mit der Konsequenz einer höheren Wahrscheinlichkeit, an SAD zu erkranken, ist spekulativ und konnte noch nicht nachgewiesen werden.
Zu 5.: Genetische Zusammenhänge
Es gibt vereinzelt Untersuchungen über genetische Einlüsse bei einer saisonal abhängigen Depression. Kathryn Roecklein und Dr. Kelly Rohan erwähnen in ihrer Übersicht aus dem Jahre 2005 Studien, die einen monogenetischen Zusammenhang mit der schon erörterten Serotonintransporter-Region 5‑HTTLPR (→ Abschnitt 4.7.5) und dem Serotonin-2A-Rezeptor-Gen untersuchten (Quelle: Kathryn A. Roecklein, Kelly J. Rohan PhD, Seasonal Affective Disorder, An Overview and Update, Psychiatry, 1/2005, 2 (1), S. 20 ‑ 26, Edgemont/Pennsylvania, USA 2005), http://www.ncbi.nlm.nih.gov/...PMC3004726/#B29).
Schon vergleichbare Untersuchungen zur asaisonalen Depression ergaben keine Hinweise für einen monogenetischen Zusammenhang und das ist auch hier der Fall. Roecklein und Rohan kommen zu der Schlussfolgerung, „dass SAD wahrscheinlich das Ergebnis eines komplexen Zusammenspiels zwischen umweltbedingten, biologischen und psychologischen Faktoren ist. Daher ist eine interdisziplinäre Forschung erforderlich, um wissenschaftliche Erkenntnisse über die Ursachen und Behandlung von SAD zu erlangen und die Gründe für die allgemein dokumentierte Zunahme von SAD bei Frauen zu verbessern.“.
Darüber hinaus ist immer noch nicht bewiesen, dass es überhaupt einen kausalen Zusammenhang zwischen einem angebllchen Serotoninmangel und der Affektregulation gibt. Und da vieles dafür spricht, dass es einen solchen Zusammenhang überhaupt nicht gibt, wären auch Untersuchungen zu Serotonin im Zusammenhang mit SAD ohne Sinn (→ Abschnitte 1.1 f.).
Es ist jedoch davon auszugehen, dass polygenetische Dispositionen bei der SAD bzw. Lichtmangeldepression von Bedeutung sind, so wie das auch schon bei der asaisonalen Major Depression festgestellt wurde. Auch Roecklein und Rohan erwähnen in ihrer Arbeit „biologische Faktoren“. Ein Beweis für polygenetische Einflüsse konnte leider noch nicht erbracht werden.
Quintessenz: Wie bei der asaisonalen Depression gibt es auch bei der SAD keine Belege für einen monogenetische Ursache. Es spricht aber vieles für polygenetische Einflüsse, jedoch sind dies derzeit lediglich Vermutungen, ein Beweis wurde noch nicht erbracht.
Zu 6.: Reaktive Aspekte
Dass viele Menschen die lang anhaltende Dunkelheit im Spätherbst und Winter stresst, muss nicht weiter ausgeführt werden. Eventuell daraus resultierende leichtere Stimmungseinbrüche haben jedoch nichts mit einer SAD oder Lichtmangeldepression im eigentlichen Sinne zu tun. Für depressive Verstimmungen während der lichtarmen Jahreszeiten wurde der Begriff des Winter‑Blues kreiert.
Es besteht theoretisch die Möglichkeit, dass sich eine bestehende Affekterkrankung durch zusätzliche jahreszeitlich bedingte depressive Verstimmungen verstärkt oder eine kurz vor dem Ausbruch stehende ernste Erkrankung überhaupt erst ausgelöst wird.
Quintessenz: Der depressiven Verstimmungen während der dunklen Jahreszeiten sind nicht mit einer echten klinisch relevanten Depression zu verwechseln. Über die Auswirkungen dieses sogenannten „Winter-Blues“ auf eine (potentielle) Depression sind dem Autoren keine Untersuchungen bekannt.
Fazit: Lichtmangel und Affektive Störungen
Der Mangel an Tageslicht steht im Verdacht, ausschlaggebender Auslöser einer saisonal abhängigen Depression (SAD) zu sein. Es gibt zahlreiche Studienergebnisse, die auf einen Zusammenhang schließen lassen, darunter auch Untersuchungen über die Wirksamkeit von Lichttherapien. Dafür können mehrere Gründe infrage kommen.
Die Vitamin‑D3‑Produktion in der Haut ist direkt vom UV‑B‑Anteil des Tageslichts abhängig, und die D3‑Synthese sinkt daher in den lichtarmen Jahreszeiten. Da D3 auch im Zentralnervensystem wichtige Aufgaben übernimmt, könnte sich das bei einem Mangel negativ auf die Steuerung und Verarbeitung von Affekten auswirken. Forschungen kamen zu dem Ergebnis, dass es einen Zusammenhang zwischen einem niedrigen D3‑Spiegel und einer Depression gibt, wobei bisher nicht bewiesen werden konnte, ob eine solche Beziehung einen kausalen Charakter hat.
Einige wenige Untersuchungen wollen einen Zusammenhang zwischen Licht und dem Serotoninstoffwechsel nachweisen, die jedoch aufgrund ihres sehr einfachen Studien-Designs nur geringevident und damit wenig beweiskräftig sind. Erkenntnisse über physiologische Vorgänge ergaben sich nicht. Darüber hinaus ist die Beteiligung des Serotonins an der Affektverarbeitung unklar und Hinweise über einen generellen Kausalzusammenhang zwischen Serotonin und einer affektiven Erkrankung gibt es bis heute nicht.
Die Produktion des Epiphysenhormons Melatonin wird bei einsetzender Dunkelheit um das 10fache gesteigert. Melatonin dämpft verschiedene Körperfunktionen. Die direkten Auswirkungen von Melatoningaben wurden in wenigen Studien untersucht. Auch diese Studien sind aufgrund ihres Designs nur als geringevident einzustufen. Interessant ist trotzdem, dass sich ein umgekehrter Zusammenhang ergab: Die Einnahme von Melatonin soll die depressive Symptomatik leicht verringert haben. Melanopsin, eigentlich ein Photopigment der Netzhaut, reguliert die Melatoninsynthse in der Epiphyse. Es gibt Hinweise, dass das Protein im Gehirn auch als gewöhnlicher Botenstoff wirkt, aber auch das konnte noch nicht zweifelsfrei bewiesen werden.
Auch eine allgemeine Jahreszeitenfehlanpassung kommt als Ursache einer SAD in Betracht. Eine potentiell durch Lichtmangel in den lichtarmen Jahreszeiten ausgelöste Reduzierung von Körperfunktionen soll zu einer gewünschten Reduktion der Leistungsfähigkeit des gesamten Organsimus führen, die von den heutigen Leistungsgesellschaften aber permanent missachtet wird. Einzelne Personen könnte das überfordern und langfristig stressbedingt eine Affekterkrankung auslösen oder zumindest daran beteiligt sein, wobei diese Hypothese ebenfalls unbewiesen ist.
Monogenetische Zusammenhänge konnten bisher ‑ wie bei der asaisonalen Depression ‑ nicht belegt werden. Es ist jedoch wahrscheinlich, dass bei der SAD ‑ analog zur asaisonalen Depression ‑ polygenetische Ursachen eine Rolle spielen.
Leichte Missstimmungen während der lichtarmen Jahreszeiten, der sogenannte Winter‑Blues, sind isoliert betrachtet eine völlig normale Reaktion. Eine schon bestehende, klinisch relevante Depression oder eine, die kurz vor dem Ausbruch steht, könnte dadurch eventuell jedoch verstärkt oder überhaupt erst ausgelöst werden. Einen Beweis gibt es auch hier nicht.
Insgesamt ist der derzeitige Wissensstand über die Zusammenhänge zwischen Lichtmangel und einer klinisch relevanten Depression bzw. SAD unbefriedigend. Ein ‑ auf welche Weise auch immer ‑ bestehender Zusammenhang erscheint zmindest plausibel. Auch aufgrund dessen befürworten Wissenschaftler und Mediziner, mehr Ressourcen in deren Erforschung zu investieren.
4.13.2 Nicht-ionisierende Mobilfunkstrahlung ▲
Mobilfunktechnik
Zur drahtlosen Übertragung von Sprachen und Daten nutzt man in der Mobilfunktechnik nicht‑ionisierende hochfrequente elektromagnetische Funkwellen, mit deren Hilfe die Informationen zwischen dem Mobiltelefon und einer Mobilfunkbasisstation ausgetauscht werden. Elektromagnetische Funkwellen übertragen neben Informationen auch Energie.
Nicht-ionisierende Strahlung hat ‑ im Vergleich zur ionisierenden Gamma‑ oder Röntgenstrahlung ‑ wesentlich weniger Energie. Sie ist daher nicht in der Lage, die Atome bzw. Moleküle der Stoffe, die sie durchdringt, zu ionisieren und neutrale Atome oder Moleküle in einen elektrisch geladenen Zustand zu versetzen.
Für die Mobilfunknetze GSM, UMTS oder LTE werden Frequenzen verwendet, die zwischen 800 und 2.600 Megahertz liegen. Ein Hertz entspricht einer Schwingung pro Sekunde.
Nachweisbare und vermutete Wirkungen elektromagnetischer Funkwellen bei Menschen
Wenn Mobilfunkwellen auf Gegenstände treffen, werden sie größenteils reflektiert und nur eine kleine Menge Energie vom Gegensstand absorbiert. Die absorbierte Energiemenge wird mit dem Messwert SAR (Spezifische Absorptionsrate) bestimmt und beurteilt.
Menschen nehmen den energetischen Teil der Funkwellen über Haut und Körpergewebe auf und wandeln ihn dort in Wärme um. Hier spricht man von thermischer Wirkung elektromagnetischer Funkwellen.
Um die Gesundheit der Menschen zu schützen und eine zu große Erwärmung zu vermeiden, wurden SAR‑Grenzwerte für Mobiltelefone und Mobilfunkbasisstationen festgelegt. Der SAR-Grenzwert für Mobilfunkstationen beträgt 0,08 Watt/kg, der für Mobiltelefone 2,0 Watt/kg (Stand 3/2015).
Neben thermischen Wirkungen werden auch athermische Wirkungen der Mobilfunkstrahlung diskutiert, die nicht mit einer Temperaturerhöhung erklärt werden können.
Zusammenhänge zwischen Mobilfunkstrahlung und Krebserkrankungen oder Auswirkungen auf die Gehirnaktivitäten werden in Erwägung gezogen. Letzters könnte Kopfschmerzen, Schlafprobleme, Schwindelgefühle, Konzentrationsschwäche oder Affektstörungen durch Mobilfunknutzung erklären.
Laut Informationszentrum Mobilfunk beträgt die örtliche Erwärmung des Gehirns während des Telefonierens etwa 0,1 Grad Celsius, die Erwärmung des Körpers durch Mobilfunkbasisstationen bei einem Abstand von wenigen Metern bereits nur 0,02 Grad Celsius. Das Informationszentrum schließt gesundheitliche Beeinträchtigungen durch thermische Wirkungen bei diesen niedrigen Werten aus und verweist auf ensprechende wissenschaftliche Untersuchungen (Quelle: Informationszentrum für Mobilfunk, Berlin, http://www.izmf.de).
Auch bezüglich potentieller athermischer Wirkungen sieht das Informationszentrum Mobilfunk keine Anhaltspunkte für schädigende Wirkungen. Es verweist auf fehlende wissenschaftlich fundierte Theorien und auf Probleme der Messbarkeit von Effekten an der Grenze zur Nachweisbarkeit (Quelle: siehe oben).
Das Bundesamt für Strahlenschutz (BfS), die Strahlenschutzkommission (SSK) und die Bundesnetzagentur gehen ebenfalls von einer Unbedenklichkeit des Mobilfunks aus. Das BfS verweist jedoch auf fehlende Beurteilungen der Langzeitwirkungen und gibt auf seiner Internet-Seite Tipps zur Reduzierung der Strahlenbelastung (Quellen: Experten-Anhörung im Bayerischen Landtag, Artikel vom 3.7.2012, diagnose:funk, Stuttgart, http://www.diagnose‑funk.org/... und Empfehlungen des BfS zum Telefonieren mit dem Handy, Bundesamt für Strahlenschutz, https://www.bfs.de/...).
Auf der Internetseite des BfS werden SAR‑Werte von Mobiltelefonen der meisten Hersteller ‑ ohne Gewähr ‑ veröffentlicht. Die Bundesnetzagentur informiert mit ihrer EMF‑Datenbank über die elektromagnetischen Felder aller Mobilfunkbasisstationen in Deutschland (Quellen bzw. Links: http://www.bfs.de/... für die Prüfung eines Mobiltelefons und http://emf3.bundesnetzagentur.de/... für die Prüfung einer Mobilfunkbasisstation).
Kontroverse Diskussionen um potentiell schädliche Mobilfunkwirkungen
Ein Blick ins Internet zeigt: Befürworter und Gegner des Mobilfunks liefern sich seit den Anfängen dieser Technik einen Schlagabtausch, jede Seite verweist auf Studien, welche die jeweilige Meinung untermauern.
So gehen alle bisher genannten Organisationen (Informationszentrum Mobilfunk, BfS und die Bundesnetzagentur) von der Unbedenklichkeit des Mobilfunks für die Gesundheit aus. Einige davon können aber nicht uneingeschränkt als neutral angesehen werden. So wurde das Informationszentrum Mobilfunk e. V. von Telefongesellschaften gegründet. Der Bund ist durch den Lizenzverkauf ebenfalls ein Nutznießer der Mobilfunktechnik.
Wie argumentieren demgegenüber Mobilfunkgegnern? Diese berufen sich häufig auf Untersuchungsergebnisse und Expertisen von Lebrecht von Klitzing, dem früheren Leiter des medizinischen Forschungslabors der Universität Lübeck. Von Klitzing gilt als einer der bekanntesten Mobilfunkkritiker in Deutschland. Er bietet heute mit seiner Firma Umweltphysik Medizinphysik Dr. Lebrecht von Klitzing verschiedene Dienstleistungen in diesem Bereich an, unter anderem auch Messungen zur Elektrosensitivität (http://www.umweltphysik.com).
In einem Interview machte von Klitzing schon 2001 folgende Aussagen: „Es gibt von wissenschaftlicher Seite bereits sehr viele Hinweise auf Gefahren ‑ auch aus Laborversuchen, bei denen Testpersonen deutlich auf die Strahlung reagierten. Die meisten Erkenntnisse haben wir von den Menschen, die in der Nähe von Mobilfunk-Basisstationen wohnen und gesundheitliche Auffälligkeiten zeigen. Diese Symptome müssen in ihrer ganzen Brandbreite ernst genommen werden (...).“ (Quelle: Strahlenexperte Klitzing im Interview: „Handys können die Gesundheit gefährden“, Spiegel-Online Wissenschaft 4/2001, http://www.spiegel.de/...126681.html).
Die im Interview genannte Messung der Elektrosensitivität verkauft von Klitzing als Testverfahren für Einzelpersonen. Von Klitzings Ansichten über die Mobilfunktechnik und sein sogenanntes ES-Testverfahren kritisieren Fachleute aus zwei Gründen: Zum einen könne er wegen seiner kommerziellen Interessen nicht die notwendige fachliche Neutralität wahren, zum anderen sei das ES-Testverfahren nicht methodenvalidiert (→ Anmerkungen in der nachfolgend zitierten Literaturstudie von Norbert Leitgeb).
Eine weitere mobilfunkskeptische Argumentation ist auf der Kritikerseite zu Mobilfunk & Elektrosmog zu finden, die von der Initiative Informationszentrum gegen Mobilfunk betrieben wird. Dort beruft man sich im Beitrag „Mit den Masten kommen die Depressionen“ auf einen Report der DAK Gesundheit aus dem Jahre 2005, welcher der Bundeshauptstadt Berlin einen höheren prozentualen Anteil an Krankheitstagen aufgrund von depressiven Störungen bescheinigt. In dem Beitrag der Kritikerseite wird dann eine Verbindung mit der hohen Anzahl der Berliner Mobilfunkbasisstationen hergestellt und behauptet, dies sei eine Erklärung dafür. Auch ein (fehlerhafter) Link zum betreffenden DAK-Report wird angeboten (Link: https://izgmf.de/Aktionen/...). In dem DAK-Report wird aber gar kein Zusammenhang mit Mobilfunkmasten hergestellt, dort wird das Wort Mobilfunk noch nicht einmal erwähnt (Quelle: DAK Gesundheitsreport 2005, DAK Versorgungsmanagement, Hamburg 2005, http://www.vaeter‑aktuell.de/studien/...).
Neben dem eher fragwürdigen Versuch, mit Hilfe der Veröffentlichung einer Krankenkasse einen nicht nachgewiesenen Kausalzusammenhang zu suggerieren, ist auch die Konstruktion eines solchen Zusammenhangs aufgrund einer ungeeigneten Zahlengrundlage fragwürdig.
Insgesamt scheint die Berichterstattung sowohl von Befürwortern als auch Gegnern des Mobilfunks eher tendenziös, wenig erhellend oder übertrieben. Für Menschen, die sich neutral informieren möchten, also nicht vertrauenserweckend.
Unabhängige Auseinandersetzungen mit diesem Thema versprechen die Untersuchungen von Norbert Leitgeb (TU Graz), der verschiedene Untersuchungen über mögliche Einflüsse der Mobilfunkstrahlung auf Hirnaktivitäten, Schlafqualität und Wohlbefinden analysiert hat, einschließlich der Durchführung einer eigenen Schlafstudie und eine Studie von Roland Glaser, zur Zeit Professor Emeritus an der Berliner Humboldt-Universität, der den Erkenntnisstand zu möglichen gesundheitlichen Auswirkungen des Mobilfunks zusammenfasste und bewertete (Quellen: N. Leitgeb, Untersuchung der Schlafqualität bei elektrosensiblen Anwohnern von Mobilfunk-Basisstationen unter häuslichen Bedingungen, Institut für Krankenhaustechnik mit Europaprüfstelle für Medizinprodukte, TU Graz/Erzherzog-Johann-Universität, in: Reihe Umweltpolitik des Bundesministeriums für Umwelt, Naturschutz und Reaktorsicherheit, Bonn 2007, http://www.bmub.bund.de/.../schriftenreihe_rs709.pdf und R. Glaser, Darstellung und Bewertung des wissenschaftlichen Erkenntnisstandes zu möglichen gesundheitlichen Auswirkungen des Mobilfunks in Relation zu bestehenden Empfehlungen und Normen, Oktober 2000, http://www.puls217.de/Glaser.pdf).
Die Studie von Norbert Leitgeb umfasst zwei Teile. Der erste Teil ist eine Literaturstudie, in der er sowohl sechs epidemiologische Studien als auch Labor- und EEG-Studien (11 bzw. 26 Studien) bewertete. Er stellte fest, dass die untersuchten epidemiologischen Studien in fünf von sechs Fällen mit zum Teil erheblichen methodischen Schwächen durchgeführt wurden, eine Studie erbrachte keine Nachweise zwischen Mobilfunknutzung und Schlafstörungen. Mit Blick auf Laborstudien bemängelte er generell deren methodische Nachteile, weil die Teilnehmer beispielsweise in einer für sie fremden Umgebung schlafen müssen oder Labore nicht in der Lage seien, reale Bedingungen ausreichend exakt nachzubilden.
In seiner Untersuchung bewertete er auch Studien des schon erwähnten Mobilfunkkritikers Lebrecht von Klitzings. Dazu heißt es: „Klitzing (1992) berichtet über Veränderungen der α‑Leistung des EEGs aufgrund von Untersuchungen in 5 fest vorgegebenen Intervallen von je 15min Dauer bei Exposition gegenüber mit 217Hz gepulsten 150MHz‑Feldern im jeweils 2. und 4. Intervall, jedoch ohne Angaben über die statistische Signifikanz. Meckelburg et al. (1995) versuchten, diese Ergebnisse in einer nachfolgenden Studie unter Mitwirkung von Klitzings zu replizieren, konnten sie jedoch nicht bestätigen. Bei der Auswertung der EEGs ergaben sich Hinweise, dass die berichteten Veränderungen der α‑Leistung auf Vigilanzänderungen während der langen Versuchsdauer zurückzuführen sein dürften. Klitzing (1995) exponierte 17 Probanden gegenüber einer Strahlung von 150MHz, die mit 217Hz gepulst war, mehrmals innerhalb von 15 Minuten. Nach der ersten Exposition kam es zu einer deutlich erhöhten Amplitude im Alpha‑Band, was jedoch in der Arbeit nicht statistisch belegt wurde.“.
In seiner Zusammenfassung der Studienbewertungen stellte Leitgeb fest: „In der Mehrheit der Studien wird über einen Einfluss der Exposition durch EMF [elektromagnetische Felder, Anmerkung des Verfassers], wie sie von Mobiltelefonen ausgesendet werden, berichtet. Die statistischen Signifikanzen sind jedoch unterschiedlich. Es wurde in keiner Studie eine Bonferoni-Korrektur des Ergebnisses durchgeführt. Da die Expositionsbedingungen jedoch sehr unterschiedlich sind, fehlen darüber hinaus Bestätigungen durch unabhängige andere Gruppen. Die bisherigen Ergebnisse sind daher lediglich als Hinweise auf ein mögliches Auftreten spektraler Veränderungen anzusehen, das zur Exposition zeitlich verzögert sein und über die Expositionszeit hinaus anhalten kann. Aufgrund der vorliegenden Studien kann eine mögliche Beeinflussung des Wach-EEGs durch EMF von Mobiltelefonen nicht ausgeschlossen werden. Zwei von den drei Arbeiten zu auditiv evozierten EEG-Potentialen zeigten einen möglichen Einfluss von EMF von Mobiltelefonen. Die Untersuchungsergebnisse zu visuell evozierten Potentialen und sonstigen durchgeführten Tests zur Gehirnaktivität sind widersprüchlich. In Bezug auf die Schlafstudie weisen die Ergebnisse auf die Wichtigkeit hin, die Rohdaten der EEG-Registrierungen für weitere Auswertungen verfügbar zu haben, da die berichteten subtilen Effekte aus den software-generierten Schlafparametern nicht ableitbar sind. Ob Elektrosensibilität im Sinne einer kausalen Beziehung zu elektromagnetischen Feldern besteht, ist nach wie vor nicht belegt. Es zeigte sich jedoch, dass die Strategie zur Rekrutierung der Probanden einen Einfluss auf das Untersuchungsergebnis hat.“.
Im zweiten Teil führte Norbert Leitgeb selber eine Schlafuntersuchung durch, bei der nicht - wie sonst üblich - Reaktionen auf erzeugte elektromagnetische Strahlung in einem Labor untersucht wurden, sondern man wesentlich realitätsnäher am Ort der Schlafprobleme - also in den Schlafzimmern der Probanden - vorhandene Immissionen abschirmte. Das Studienziel bestand darin zu prüfen, ob Effekte überhaupt nachweisbar sind - und zwar unter speziell ausgewählten Personen, die der Meinung sind, sie seien elektrosensibel und unter starkem Leidensdruck stehen, das heißt unter besonders häufigen und massiven Schlafstörungen litten. Bei einem negativen Studienergebnis könnte man darauf schließen, dass auch bei weniger empfindlichen Personen keine Auswirkungen zu erwarten sind. Von den etwa 600 infrage kommenden Probanden wurden 97 in die Studie eingeschlossen, aufgrund von Absagen und Abbrüchen führten letztlich 44 Personen die Studie komplett durch.
In 3 x 3 Untersuchungen verbrachten die Teilnehmer die Nächte jeweils unter einem „echten“ Verum-Schirm, unter einem die elektromagnetische Strahlung nicht beeinflussenden Sham-Schirm (Placebo) und zur Kontrolle ohne Abschirmung, wobei die Immissionen zur Qualitätskontrolle innerhalb des Schirms überwacht wurden. Außerdem gab es noch eine „Eingewöhnungsnacht“ ohne Datenauswertung. Die Teilnehmer hatten weder Informationen über die Verwendung unterschiedlicher Schirme noch waren diese aufgrund ihrer Textilstruktur unterscheidbar. Die Messerfassung und Messauswertung erfolgte durch unterschiedliche Teams, die Versuchsreihenfolgen waren dem auswertenden Team nicht bekannt und konnten auch nicht bestimmten Personen zugeordnet werden (Doppelblind-Studie). Es wurden subjektive (Befragungen) und ojektive Parameter (Messungen) einbezogen.
In der Zusammenfassung der Auswertungen der Schlafstudie schreibt Leitgeb: „(...) Als neue Alternative zu Provokationsstudien wurden mit einem mobilen Schirm die vorhandenen Felder in den Schlafzimmern der Probanden abgeschirmt und die Schlafqualität der Probanden mit und ohne Schirm verglichen, um die potenzielle Rolle des hochfrequenten 'Elektrosmogs' untersuchen zu können. Dabei war es entscheidend, erkennen zu können, ob bereits der Glaube an die Wirksamkeit des Schirmes eine Verbesserung oder ob die Beeinträchtigung durch die ungewohnte Umhüllung des Bettes mit einem Schirmstoff eine Verschlechterung der Schlafqualität bewirkt. Dies wurde dadurch ermöglicht, dass ein ununterscheidbarer zweiter Schirm verwendet wurde, mit dem eine 'Elektrosmog'-Abschirmung lediglich vorgetäuscht wurde. Es konnten daher drei Untersuchungsbedingungen (Verum / Sham / Kontrolle) während mindestens je drei Nächten ausgewertet werden. Durch Anwendung univarater [sic!] und multivariater statistischer Analysemethoden konnte gezeigt werden, dass aufgrund des randomisierten Untersuchungsansatzes keine signifikanten Beeinflussungen der Schlafanalyse durch Kofaktoren aufgetreten sind. Eine wesentliche Voraussetzung für belastbare Ergebnisse war, dass die Untersuchungen unter Doppelblind-Bedingungen durchgeführt wurden und dass insbesonders auch die Probanden nicht wussten, wann die Elektrosmogabschirmung echt und wann nur vorgetäuscht war. Zur Qualitätssicherung waren im Versuchsdesign einige dies bezügliche Kontrollmöglichkeiten vorgesehen worden. (...) Obwohl es kein Auswahlkriterium war, zeigte es sich, dass die meisten Probanden eine gegenüber der Allgemeinbevölkerung erhöhte Elektrosensitivität aufwiesen. Die Messungen der elektromagnetischen Immissionen haben die Erwartungen der Probanden nicht bestätigt, wonach in ihren Schlafzimmern atypisch hohe elektromagnetische Immissionswerte festzustellen sein würden. Die gepoolte Auswertung für die Gesamtgruppe aller Probanden ergab einen statistisch nicht signifikanten Trend zu einer Verbesserung der subjektiv empfundenen Schlafqualität mit zunehmender Immissionsstärke. Auch bezüglich des vom Mobilfunk stammenden Immissionsanteils ergab sich kein signifikanter Zusammenhang. Mit Hilfe einer neu entwickelten Darstellungsmethode, des Effekt-Quadranten-Diagramms, konnte eine Zusammenschau der Einzelergebnisse vorgenommen und alle drei Untersuchungsbedingungen gemeinsam sowohl für einzelne Schlafparameter als auch für einzelne Probanden bewertet werden. Bei der überwiegenden Anzahl der Probanden konnte die subjektive Überzeugung widerlegt werden, dass Elektrosmog an den Schlafstörungen schuld sei. Die gepoolte Auswertung ergab statistisch signifikante Placebo-Effekte bei subjektiven Schlafparametern. Dies wurde durch Probanden-spezifische Auswertungen bestätigt. Bei 18 Prozent der Probanden konnte nachgewiesen werden, dass bereits der Glaube an eine Schirmwirkung zu einer Schlafverbesserung führte (Placebo-Effekt). Bei 7 Prozent der Probanden konnte der Nachweis erbracht werden, dass sie sich über die Versuchsbedingungen Klarheit verschafft hatten. Ihre Ergebnisse mussten daher für die Untersuchung einer potentiell kausalen Wirkung der hochfrequenten Immissionen ausgeschieden werden. Bei 9 Prozent der Probanden konnte eine statistisch signifikante Beeinflussung des Einschlafverhaltens durch die Verringerung der hochfrequenten Immissionen festgestellt werden. Die Ergebnisse waren innerhalb der Gruppe konsistent (d. h. es ergaben sich bei keinem Probanden gegenteilige Veränderungen) und wurden durch ergänzende an der Signifikanzgrenze liegende Befunde gestützt. Demnach ergaben sich bei allen signifikanten Ergebnissen der Gruppe bei Verringerung des 'Elektrosmogs' verlängerte Einschlafzeiten. Dies ist in Übereinstimmung mit Ergebnissen in der Literatur, wonach sich bei Verstärkung hochfrequenter Immissionen Verkürzungen der Einschlafzeiten ergaben. Die Untersuchungen ergaben keinen Hinweis auf gesundheitsschädigende Einflüsse der hochfrequenten elektromagnetischen Immissionen auf den Schlaf, insbesonders auch nicht durch jene des Mobilfunks. Die Studie zeigte jedoch, dass subtile Auswirkungen hochfrequenter Immissionen auf das Einschlafverhalten möglich sind. Dieses Ergebnis ist insbesonders in Hinblick auf die Diskussion über mögliche Wechselwirkungsmechanismen schwacher (nichtthermischer) hochfrequenter elektromagnetischer Felder von Bedeutung und sollte daher durch weitere Untersuchungen abgesichert werden.“.
In seiner Darstellung und Bewertung des Erkenntnisstandes zu möglichen gesundheitlichen Auswirkungen des Mobilfunks setzte sich der Autor Roland Glaser mit 459 wissenschaftlich rezensierten Studienpublikationen auseinander. Glaser bewertete die Zuverlässigkeiten von Methodik, Datenerhebung und Ergebnisinterpretation und verfolgte damit ein ähnliches Ziel wie ca. sechs Jahre später Norbert Leitgeb. Neben epidemiologischen Untersuchungen und Versuchen mit Probanden bewertete Glaser zusätzlich auch Tierversuche und Studien an molekularen und zellulären Systemen in-vitro. Auch wenn der Veröffentlichungszeitpunkt von Glasers Publikation schon länger zurückliegt, sind deren Ergebnisse und Schlussfolgerungen auch heute noch relevant.
Glaser weist auf die mangelnde Aussagekraft epidemiologischer Langzeit-Studien hin, denn diese können nicht annähernd die hohen Anforderungen erfüllen, die an Studienparamenter zu stellen sind. In-vitro-Experimente eignen sich nach seiner Auffassung nur bedingt zur Beurteilung gesundheitlicher Auswirkungen auf den gesamten Organismus, mit Ausnahme genetisch relevanter Aussagen. Mit Probanden-Studien könnten zwar Kurzzeitreaktionen nachgewiesen werden, sie lassen aber keine Rückschlüsse bezüglich der Wirkungsmechanismen zu. Die Untersuchung von Langzeitreaktionen sei zwar mit Tierexperimenten möglich, die jedoch nur bedingt Rückschlüsse auf den Menschen ermöglichen.
Über festgestellte Reaktionen des Zentralnervensystems auf Bestrahlungen in Probanden-Studien schrieb Glaser: „Bemerkenswert erscheint jedoch, dass offenbar reversible Reaktionen des Gehirns auf die Einstrahlung dieser Felder nicht auszuschließen sind. Obgleich die vorliegenden Resultate nicht reproduzierbar sind, und somit nicht als abgesichert betrachtet werden dürfen, weisen sie doch auf einige Reaktionen hin. Diese Reaktionen sind reversibel und gehören in die Kategorie von alltäglichen Antworten auf verschiedenste gewollte und ungewollte Umwelteinflüsse. Im Zusammenhang mit unserer völligen Unkenntnis möglicher Mechansimen der Wirkung derart schwacher HF-Felder verdienen diese Ergebnisse jedoch Beachtung. Ähnliche Effekte, ausgelöst durch Licht- oder Schall-Einflüsse, durch Anregungsmittel, wie Coffein, Alkohol etc. weisen nach, dass die von bekannten Rezeptoren aufgenommenen Reize einer bestimmten neuronalen Verarbeitung unterliegen. Von schwachen HF-Feldern sind uns jedoch solche Rezeptoren nicht bekannt und bisher sind auch keine Mechanismen vorstellbar, welche zu einer Rezeption schwacher HF-Felder führen könnten. Eine wissenschaftlich eindeutig belegte neuronale Reaktion würde immerhin auf einen Wirkungsmechanismus schwacher Felder deuten.“.
Bei der Auswertung und Interpretation wissenschaftlicher Untersuchungen muss laut Glaser unbedingt berücksichtigt werden, dass ein experimentell nachgewiesener Effekt nicht mit einem Schaden im medizinischen Sinne gleichgesetzt werden darf.
Am Ende seiner Publikation zieht Roland Glaser u. a. folgende Schlüsse: „Generell ist festzustellen, dass es in Auswertung der wissenschaftlichen Publikationen keinen Anhaltspunkt dafür gibt, dass die derzeit im Mobilfunk verwendeten Felder (...) gesundheitliche Schäden verursachen. Diese Einschätzung basiert auf den in der Studie im Detail diskutierten Experimenten mit menschlichen Probanden und an Versuchstieren sowie auf in-vitro-Experimenten mit Zellkulturen, und bezieht sich sowohl auf akute Befindlichkeits-Störungen als auch auf Spätfolgen, wie z. B. die Krebsentstehung. Wenn auch noch nicht durch unabhängige Untersuchungen bestätigt, so liegen andererseits inzwischen einige wenige, jedoch ernst zu nehmende Befunde an Probanden vor, die 'Effekte' an Menschen in einem Dosisbereich unterhalb der Grenzwerte hinweisen (elektrophysiologische Signale, Hormon-Veränderungen). Wenn diese Veränderungen auch im Bereich alltäglicher Schwankungen und Auslenkungen liegen, vergleichbar solchen, die z. B. durch plötzliche Geräusche, optische Signale etc. ausgelöst werden oder durch alltägliche pharmakologische Beeinflussungen (z. B. durch Coffein), so weisen sie doch, falls sie sich reproduzierbar bestätigen ließen, auf bisher unbekannte biophysikalische Mechanismen der Wechselwirkung der Felder mit biologischen Systemen hin. Wenn diese Befunde auch die oben getroffene Aussage bezüglich gesundheitlicher Irrelevanz nicht in Frage stellen so ist es dennoch erforderlich, diesen Phänomenen nachzugehen und die ihnen zugrunde liegenden Mechanismen und Bezüge zu klären. Aus den derzeit verfügbaren epidemiologischen Erhebungen ergeben sich ebenfalls keine Gefahren für die Benutzer von Mobilfunkgeräten sowie für Bewohner von Gebieten in unmittelbarer Nähe von Sende-Antennen. (...) Zu mindestens bei der Benutzung eines Mobil-Telefons, weniger in der üblichen Nähe von Sendemasten, werden tatsächliche Intensitäten erreicht, die nur wenig unter den zulässigen Grenzwerten liegen. (...) Diese Umstände erfordern für den Bereich des Mobilfunks eine erhöhte Aufmerksamkeit und eine genaue Dosimetrie. (...) Beide Anforderungen treffen sich in der Notwendigkeit, Klarheit über mögliche biophysikalische Wirkungsmechanismen zu erlangen.“.
Fazit: Potentielle Auswirkungen nicht-ionisierender Mobilfunkstrahlung auf Affekte
Es spricht vieles dafür, dass von der Mobilfunkstrahlung keine unmittelbaren gesundheitlichen Gefahren für das Zentralnervensystem ausgehen ‑ weder durch thermische noch durch athermische Wirkungen.
Dennoch sind biophysikalische Veränderungen durch die Strahlung auch im Gehirn feststellbar, ohne dass bis zum jetzigen Zeitpunkt geklärt werden konnte, auf welchen Wirkungsmechanismen diese beruhen, oder ob mit ihnen langfristig gesundheitlichen Gefährdungen verbunden sind und diese ein erhöhtes Aufkommen neurologisch-psychiatrischer Erkrankungen zur Folge hätten. Die Studienergebnisse deuten jedoch eher drauf hin, dass es sich um reversible und normale Reaktionen auf einen Umwelteinfluss handelt.
Eine eindeutige und wissenschaftlich begründbare endgültige Bewertung der Mobilfunkstrahlung hinsichtlich ihrer kurz- und langfristigen Auswirkungen auf das Zentralnervensystem, Affekte und Affektstörungen ist derzeit aus diesen Gründen nicht möglich.
4.13.3 Ionisierende Strahlung ▲
Ionisierende Strahlung ist ein Sammelbegriff verschiedener Strahlenarten ähnlicher Eigenschaften, wobei Alpha-, Beta-, Gamma-, Röntgen- und Protonenstrahlung unterschieden werden.
Alpha-, Beta- und Gamma-Strahlen werden auch als radioaktive Strahlen bezeichnet. Sie entstehen durch zerfallende Atomkerne (= Radionuklide) meist natürlich vorkommender Elemente, deren instabilen Kerne mehr Neutronen als Protonen enthalten und Strahlung emittieren. Der Fachbegriff für ein Element mit derartigen Eigenschaften lautet Isotop. Die Differenzierung in die drei Strahlengruppen Alpha, Beta und Gamma erfolgt aufgrund unterschiedlicher Materiedurchdringungsfähigkeiten der Strahlen.
Natürliche Quellen dieser Strahlenarten sind kosmische Strahlung und irdisches Gestein. Große Strahlenmengen entstehen künstlich bei der Energiegewinnung in Kernreaktoren und im Zusammenhang mit der Herstellung und Anwendung von Kernwaffen. Darüber hinaus werden Alpha-, Beta- und Gammastrahlen auch bei verschiedenen medizinischen Diagnose- und Therapienverfahren emittiert.
Röntgen- und Protonenstrahlen sind demgegenüber immer technisch erzeugte Strahlungsarten, die hauptsächlich in der Medizin zum Einsatz kommen.
Körperdurchdringende Eigenschaften ionisierender Strahlen
Die Alpha-Strahlung ist eine Teilchenstrahlung, die aufgrund ihrer großen Masse von außen nicht in den Körper eindringen kann. Lediglich die obere Schicht der Epidermis und deren tote Hautzellen sind betroffen. Demgegenüber sind mit der Nahrung oder über das Atemsystem in den Körper eingedrungene Alpha-Strahlung emittierende Substanzen für Gewebe und DNA sehr gefährlich. Hier ist vor allem das Element Radon bzw. dessen Isotope zu nennen.
Die Beta-Strahlung ist ebenfalls eine Teilchenstrahlung, die jedoch von außen etwas tiefer in die Haut vorzudringen vermag, deren schädliche Wirkung aber auf diese oder die Augenoberfläche begrenzt bleibt. Die Folgen sind Verbrennungen, Linsentrübung oder als Spätfolge Hautkrebs. Auch hier gilt: Beta-strahlende Isotope innerhalb des Körpers können große Schäden verursachen, beispielsweise Schilddrüsen- oder Knochenkrebs und Leukämie.
Bei der Gamma-Strahlung handelt es sich um eine hauptsächlich von zerfallenden Urankernen ausgehende elektromagnetische Strahlung, die aus Photonen und nicht aus Teilchen besteht. Die Gamma-Strahlung hat daher keine Masse und dringt sehr gut von außen in das Körpergewebe ein. Durch ihre Fähigkeit, Moleküle aufzubrechen, verursacht sie erhebliche Zellschädigungen. Dabei ist sowohl Gewebe als auch das Erbgut einer Zelle betroffen. Die Folgen können die tödliche Strahlenkrankheit und die Bildung von Krebszellen sein.
Trotz einiger übereinstimmender Merkmale ist die weniger energiereiche elektromagnetische Röntgenstrahlung von der Gamma-Strahlung zu unterscheiden. Die ebenfalls ionisierende Röntgenstrahlung hat ihren Ursprung nicht in zerfallenden Atomkernen, sie entsteht künstlich, vergleichbar mit dem sichtbaren Licht aus einer Glühbirne. Ungeschützt dringt Röntgenstrahlung tief ins Körpergewebe ein und schädigt insbesondere das Erbgut der Zellen.
Bei der Protonenstrahlung handelt es sich um künstlich hochbeschleunigte Protonen, die für den punktgenauen Beschuss von Tumoren in der Krebsmedizin genutzt wird. Sie ist daher vergleichbar mit der Alpha- oder Beta-Strahlung. Das auch als Partikeltherapie bezeichnete Verfahren soll gewebeschonender als herkömmliche Bestrahlungsverfahren sein. Die Partikeltherapie ist in Deutschland noch relativ unbekannt und wird aufgrund des hohen technischen Aufwands selten durchgeführt. Aufgrund dessen und ihrer prinzipiellen Ähnlichkeit mit den herkömmlichen Bestrahlungsverfahren wird hier auf eine genauere Darstellung verzichtet. Alle Erkenntnisse und Schlussfolgerungen über Bestrahlungsnebenwirkungen gelten indes auch für dieses Verfahren.
Ionisierende Strahlen verursachen Zellgewebeschäden durch Ionisation und ROS
Aggressive ionisierende Strahlen verändern molekulare bzw. atomare Strukturen. Dies tun sie entweder direkt am Zellgewebe mittels Ionisation, bei der aggressiv-zellschädigende freie Radikale entstehen, oder indirekt durch reaktive Sauerstoff-Spezies (ROS, Abkürzung des englischen Fachterminus Reactive Oxygen Spezies).
ROS sind reaktionsfreudige Verbindungen, die in einem Zusammenhang mit Sauerstoff stehen und werden ‑ abgeleitet vom lateinischen Wort Oxygenimum für Sauerstoff ‑ Oxidantien genannt. Sie bestehen aus zwei Gruppen aggressiver Substanzen: freien Sauerstoffradikalen und nicht‑radikalen reaktiven Sauerstoffverbindungen und entstehen durch natürliche endogene Zellprozesse ‑ insbesondere durch die natürliche Zellatmung ‑, aber eben auch durch ionisierende Strahlen, die beispielsweise Wasser (H2O) durch die Wasserradiolyse verändern. Für eine Zelle, die zu 80% aus Wasser besteht, ist dieser Mechanismus brandgefährlich.
Ionisation und ROS haben das Potential, sämtliche Zellgewebearten zu schädigen, einschließlich der wichtigen Zellmembranen und der DNA. Der Anteil direkter Strahlenschädigung am Gewebe durch Ionisation beträgt ca. 30 bis 40%, der Anteil der Schädigung durch alle ROS ‑ d. h. durch ionisierende Strahlung und andere Prozesse ‑ beträgt ca. 60 bis 70% (Quelle: Patricia Virsik-Köpp, Biologische Strahlenwirkungen, Zentrum Radiologie, Universitätsmedizin Göttingen).
Die Entstehung freier Radikaler und ROS auf atomar-molekularer Ebene
Zwei Mechanismen führen bei ionisierender Strahhlung zu freien Radikalen bzw. ROS:
- Die Strahlen brechen die Bindungen zwischen zwei beliebigen Atomen eines Moleküls. Es entstehen zwei Molekülteile bzw. Molekülreste. An beiden Molekülresten befindet sich jetzt außen jeweils ein Atom mit einem ungepaarten Elektron. Aus dem vorher neutralen und funktionsfähigen Biomolekül sind kurzfristig zwei biologisch funktionslose ‑ aber stattdessen potentiell schädliche ‑ freie Radikale geworden.
- Die Strahlen können darüber hinaus ein Elektron aus neutralen Atomen oder aus Molekülen entfernen, ohne dass es zu einem Molekülbruch kommt. Es entsteht auch hier kurzfristig ein freies Radikal mit einem ungepaarten Elektron.
Direkte und indirekte Gewebeschäden durch Ionisation bzw. ROS stellen die eigentlichen Probleme ionisierender Strahlung dar, denn sie führen bei übermäßiger Präsenz in allen Gewebearten und vor allem an der DNA zu einer zerstörerischen Kettenreaktion durch Elektronenraub, denn die freien Radikale füllen nun ihre Elektronlücken. Dafür haben sie zwei Möglichkeiten:
- Freie Radikale haben ihre Bezeichnung unter anderem ihrer unangenehmen Eigenschaft zu verdanken, sich das nötige Elektron bei anderen Substanzen zu beschaffen, es also anderen Atomen oder Molekülen zu entreißen. Die vormals freien Radikale sind dann zwar wieder neutral, die biologische Funktionslosigkeit im Falle der Molekülreste bleibt aber bestehen.
Insbesondere ist der Umstand verheerend, dass sie ihre radikale Eigenschaft transferieren, denn das Opfermolekül wird jetzt seinerseits zum freien Radikal mit einem ungepaarten Elektron und der für die Zelle schädliche Prozess des Elektronenraubs geht an anderer Stelle weiter.
- Für Molekülreste besteht durch die Verbindung mit einem anderen Molekül eine zweite Möglichkeit, die Elektronlücken zu beseitigen und wieder neutral zu werden. Dies kann sowohl ein freies Radikal oder ein neutrales Mokekül sein. Wenn sich der radikale Molekülrest nicht zufällig wieder mit seinem ursprünglichen Pendant verbindet, entsteht jedoch ein neues, wiederum funktionsloses oder manchmal auch gefährliches Biomolekül.
Mittel- und langfristige Zellschäden durch ionisierende Strahlung
Konsequenzen reaktiver Sauerstoff-Spezies (ROS) werden beim Thema Zellatmung und reaktive Sauerstoff-Spezies (ROS) ausführlicher erörtert (→ Abschnitt 4.13.6). Die dort beschriebenen Prozesse treffen im Prinzip auf direkte und indirekte Gewebeschäden durch ionisiernde Strahlung zu, auch wenn einige Unterschiede bestehen. Aus Gründen der Vereinfachung wird auf die Darstellung dieser Unterschiede verzichtet. Nachfolgend daher nur eine kurze Zusammenfassung der wichtigsten Fakten über potentielle Folgen ionisierender Strahlung.
Zunächst ist festzustellen: Eine Zelle ist unter normalen Bedingungen in der Lage, freie Radikale und ROS mit ihren antioxidativen Prozessen zu neutralisieren. Schäden können entstehen, wenn sich in einer Zelle mehr freie Radikale bzw. ROS befinden, als diese neutralisieren und beseitigen kann und die antioxidativen Schutzmechnismen nicht mehr ausreichen. Dieser Zustand wird im Falle der ROS als oxidativer Stress bezeichnet. Die Folgen sind potentielle Schäden an sämtlichen Zellbestandteilen: Lipiden, Lipoïden, Enzymen/Proteinen und Nukleinsäuren (DNA).
Vor allem nicht reparierte DNA-Schäden sind gefährlich, denn sie werden als Mutationen auf nachfolgende Zellgenerationen übertragen und können zu langfristigen Problemen der Proteinbiosynthese führen. Gefährdet sind sämtliche Gene bzw Codes, also auch ncRNA-Codes (→ Abschnitt 3.3 für die Entstehung und Reparatur von DNA-Mutationen und Abschnitt 4.2 für die Mutationsfolgen). Mutationsschäden können (zunächst) unbemerkt bleiben, aber auch zu mehr oder weniger großen Zellprozessproblemen führen.
Ebenfalls ist ein Zelltod möglich, der durch eine Apoptose geregelt ablaufen kann, mit der die geschädigte Zelle gründlich und ohne weitere negative Konsequenzen für Nachbarzellen „entsorgt“ wird . Die durch ionisierende Strahlung ausgelösten Zellschäden führen aber in der Regel zu chaotisch und ungeregelt ablaufenden Zell‑Nekrosen, die meist von entzündlichen Prozessen begleitet werden und benachbartes Gewebe zusätzlich schädigen. Der Zelltod durch ionisierende Strahlen führt also häufig zu einer doppelten Belastung von Zellen und Gewebe.
Strahlenbelastung aufgrund umweltbedingter ionisierender Strahlung
Die beiden natürlichen radioaktiven Strahlungsquellen der Umwelt sind die von der Erde ausgehende terrestrische Strahlung und die kosmische Strahlung aus dem Weltraum.
Die terrestrische Strahlenbelastung basiert auf instabilen, zerfallenden Atomkernen (Radionukliden) instabiler Elemente (Isotope), die in Böden und Gesteinen als natürliche Isotope vorkommen. Für Menschen haben vor allem das Kalium-Isotop K-40, das Radon-Isotop Rn-222 und - je nach Region - auch Isotope des Urans eine Bedeutung. Bei der terrestrischen Strahlungswirkung wird zusätzlich noch nach der Außen- und Innenwirkung differenziert, also wie ionisierende Strahlen den menschlichen Körper treffen. Die äußere Komponente der Strahlung wirkt ausschließlich von außen auf den Körper ein, während die innere Komponente das Gewebe von innen über Nahrung, Tabakgenuß und Atemluft erreicht (Stand 6/2021, Quelle: Bundesamt für Strahlenschutz, Salzgitter 2015, https:///www.bfs.de/...).
Der größte Teil der kosmischen Strahlung hat seinen Ursprung in der Sonne. Die Stärke der kosmischen Strahlenbelastung ist abhängig von der Höhenlage: Menschen in höheren Wohnlagen, Vielfliegende und Raumfahrer sind im Schnitt stärker belastet.
Die Höhe der Strahlendosis (Strahlenexposition), der ein Mensch pro Jahr durch natürliche Strahlung ausgesetzt ist, wird in Millisievert bzw. Mikrosievert gemessen. Die durchschnittliche natürliche Strahlenbelastung in Deutschland liegt bei 2,1 Millisievert pro Jahr, schwankt aber - je nach Region und Lebensstil - zwischen einem und 10 Millisievert (Stand 6/2021, Quelle: Bundesamt für Strahlenschutz, Salzgitter 2015, https:///www.bfs.de/...).
Der Gesamtwert von 2,1 Millisievert setzt sich aus den nachfolgend aufgeführten Komponenten zusammen:
- Über die Atemluft und Nahrung werden Substanzen aufgenommen, die das Körpergewebe im Schnitt einer Strahlendosis in Höhe von 1,4 Millisievert aussetzen, davon gehen 1,1 Millisievert auf Radon zurück, der Rest in Höhe von 0,3 Millisievert basiert auf Kalium-40 und Uran- bzw. Thorium-Isotopen.
- 0,7 Millisievert machen die äußeren Strahlenexpositionskomponenten aus: 0,3 Millisievert durch kosmische Strahlung, 0,4 Millisievert durch terrestrische Strahlung aufgrund natürlicher radioaktiver Stoffe in Böden, Gesteinen und Baumaterialien.
Da davon auszugehen ist, dass sich alle Lebewesen auf der Erde im Laufe der Jahrmillionen an die natürliche ionisierende Strahlung angepasst haben, sind Schäden alleine durch sie nicht zu erwarten. Evolutionsforscher und Genetiker nehmen an, dass die natürliche Strahlung und darauf basierende natürliche Mutationen sogar sehr wichtig für die evolutorische Entwicklung und Anpassung der Arten auf der Erde sind.
Die Durchschnittswerte gelten nicht bei Menschen, die einer wesentlich größeren Belastung durch natürliche Strahlenexposition ausgesetzt sind als der Rest der Bevölkerung. Dabei handelt es sich um folgende Personengruppen:
- Flugpersonal bzw. Vielflieger,
- Bewohner hoher bzw. höherer Gebirgslagen,
- Bewohner ozongeschädigter Gebiete, zum Beispiel in polnahen Regionen (z. B. Australien),
- Raucher und
- Bewohner von Regionen erhöhten Radionuklidvorkommens durch Radon-, Kalium- oder Uran-Isotope.
Gefährdung und Belastung durch ionisierende Strahlung künstlicher Quellen
Neben der natürlichen Strahlung besteht aber auch noch eine allgemeine Gefährdung der Bevölkerung aufgrund einer künstlichen Strahlungsexposition durch den Betrieb technischer Anlagen sowie Fallouts. Betroffen sind:
- Kernkraftwerke,
- Forschungszentren,
- Kernbrennstoff verarbeitende Betriebe,
- Kernwaffen verarbeitende Rüstungsbetriebe,
- Atommüll-Lagerstätten,
- Uranabbaugebiete (in Deutschland die Sanierung der Wismut GmbH),
- Rückstände aus der Braun- und Steinkohleförderung,
- Sonstige industrielle Rückstände, auch aus der Erdöl- und Erdgasförderung,
- Stein- oder Braunkohleverbrennung sowie
- Fallouts nach Kernwaffentests und Kernkraftwerksunfällen.
Die Summe solcher Strahlenexpositionen wurde im Jahresbericht des Bundesumweltministeriums für Deutschland wie folgt angegeben: Jeweils < 0,01 Millisievert im Jahr für den Fallout von Kernwaffentests und die Tschernobyl-Reaktorkatastrophe, ebenfalls jeweils < 0,01 Millisievert im Jahr für kerntechnische Anlagen sowie Technik & Forschung (Quelle: Radioaktivität und Strahlung, Bayerisches Landesamt für Umwelt LfU, Augsburg 2013, http://www.lfu.bayern.de/umweltwissen/...).
Die zusätzlichen Belastungen liegen demnach weit unter der Exposition durch natürliche Strahlung, wobei es sich auch hier wieder um Durchschnittswerte handelt. So sind in den oben aufgezählten Branchen tätige Personen einer wesentlich höheren Belastung ausgesetzt. Die Stärken von Fallouts können sich wiederum von Region zu Region erheblich unterscheiden.
Über sonstige Unfälle mit radioaktiven Substanzen ist in Deutschland nichts bekannt. Im Ausland gab es in der Vergangenheit immer wieder größere Zwischenfälle, beispielsweise im Jahre 2000 auf einem Schrottplatz in der Peripherie von Bangkok/Thailand oder 1966 in Palomares/Spanien.
Ionsisierende Strahlung in medizinischen Anwendungen
Die von Radon und seinen Isotopen ausgehende Alpha-Strahlung wird in der Radontherapie genutzt, beispielsweise in Heilstollen mit radonhaltiger Luft, in denen sich Patienten aufhalten und diese Luft einatmen. Die aufgrund der damit verbundenen Strahlenbelastung umstrittene Therapie wird insbesondere bei Erkrankungen des Bewegungsapparates und des Atemsystems durchgeführt.
In geringerem Maße werden Substanzen, die Alpha-Strahlung emittieren, auch in der Behandlung von Krebs und einiger anderer Erkrankungen verwendet. So kommt bei der Radionuklidtherapie des Morbus Bechterew manchmal alpha-strahlendes Radium zum Einsatz. Dabei wird die strahlende Materie im erkrankten Gewebe maximal hoch angereichert - bei möglichst geringer Ansammlung im gesunden Gewebe.
In der überwiegenden Zahl der Radionuklidbehandlungen wird jedoch Beta‑Strahlung genutzt. Beta-Strahlen verwendet man in geringerem Umfang auch zur Durchführung der Brachytherapie (Kontaktbestrahlung), bei der kleine Strahlenquellen direkt im Körper platziert werden. In den meisten Fälle emittieren die Strahlenquellen bei der Brachytherapie jedoch Gamma-Strahlung.
Diagnostisch kommt Beta-Strahlung noch bei einer Untersuchung mit dem Positronen-Emissions-Tomographen zum Einsatz.
In der Strahlentherapie tiefer liegender Karzinome ‑ auch Teletherapie genannt ‑ ist die Gamma-Strahlung die am häufigsten verwendete Strahlungsart.
Röntgenstrahlung wird ‑ neben ihrer hauptsächlichen diagnostischen Anwendung beim Röntgen ‑ auch zur Oberflächenbestrahlung bösartiger Hauterkrankungen oder am Auge eingesetzt.
Auch hier veröffentlicht das Bundesamt für Strahlenschutz Durchschnittswerte für die Strahlenexposition. Die Belastung durch die Röntgendiagnose wird mit 1,8 Millisievert im Jahre 2009 angegeben. (Quelle: Strahlenthemen: Röntgendiagnostik, Bundesamt für Strahlenschutz, Salzgitter 2015, https://www.bfs.de/...).
Die durchschnittlichen Belastungswerte aufgrund therapeutischer Anwendungen - hauptsächlich in der Krebstherapie - sind noch wesentlich niedriger als die durch Röntgendiagnostik, was ja nicht verwunderlich ist aufgrund der vergleichsweise geringen Anzahl von Bestrahlungspatienten.
Aufgrund der Individualiät medizinisch bedingter Strahlenexpositionen sind Durchschnittswerten für den Einzelfall auch hier wenig aussagekräftig.
Ionisierende Strahlungen in der Therapie zentralnervöser Erkrankungen
Die Strahlenbelastungsdurchschnittswerte medizinischer Anwendungen haben für strahlentherapeutisch behandelte ZNS-Patienten keine Relevanz, denn hier ist die Exposition mit ionisierenden Strahlen im individuellen Fall um ein Vielfaches höher. Daher ist immer zu klären, welcher Strahlenbelastung ein Patient über die gesamte Behandlungsdauer ausgesetzt war oder sein wird.
Durch technisch weit fortgeschrittene radioonkologische Verfahren kann ein Bestrahlungsfeld heute zwar relativ exakt auf einen Tumor und das potentiell betroffene und sich in unmittelbarer Nachbarschaft befindende Gewebe begrenzt werden, es werden aber bei bestimmten Indikationen auch Bestrahlungen des gesamten Gehirns ohne Begrenzung durchgeführt.
Die absorbierte Energiedosis im Gewebe aufgrund der Bestrahlung wird in Gy (= Gray) gemessen.
Die zu verwendende Bestrahlungsstärke hängt von mehreren Faktoren ab, beispielsweise von der Strahlenempfindlichkeit der Tumorart und des umliegenden gesunden Gewebes, von der Tumorgröße oder der Sichtbarkeit des Tumors. Laut Deutscher Krebsgesellschaft sollte die absorbierte Energiedosis bei malignen Hirntumoren lokal zwischen 54 bis 60 Gy betragen. Hirnmetastasen, die sich in großen Bereichen überall im Gehirn befinden können, bekommen 30 Gy Bestrahlungsenergie, wobei hier das gesamte Gehirn bestrahlt wird (Quelle: Strahlentherapie bei Hirntumoren, Onko Internetportal der Deutschen Krebsgesellschaft e. V., Berlin, http://www.krebsgesellschaft.de/onko-internetportal/...). Solche Gesamtbestrahlungen des Gehirns werden auch prophylaktisch bei Primärkarzinomen des Körpers durchgeführt, die bekanntermaßen häufig ins Gehirn streuen.
Bestrahlungen sind immer eine starke Belastung auch für das nicht vom Tumor betroffene Gehirngewebe. Es kommt zu kurz- und langfristigen Nebenwirkungen bei Anwendung einer Strahlentherapie. Viele - besonders die stärkeren - Folgen einer Bestrahlung zeigen sich erst nach Wochen oder Monaten. Meist sind kognitive und emotionale Probleme die Folgen.
Im Allgemeinen werden in der Literatur folgende kurz- und langfristigen Bestrahlungsnebenwirkungen genannt, die auf zentralnervöse Störungen hinweisen: Übelkeit, Erbrechen, Schwindel, Störungen der Konzentrations- und Merkfähigkeit, Lernprobleme, generelle intellektuelle Einschränkungen, Erschöpfung oder Störungen der Sexualfunktion.
Die letzte Stufe einer Gewebeschädigung nach Bestrahlung, bei der Körpergewebe abstirbt, wird als Strahlennekrose bezeichnet. Die Latenzzeit derart massiver Schädigungen kann sehr hoch sein. Es wurden Fälle beschrieben, bei denen zwischen Bestrahlung und dem Untergang von Körpergewebe mehr als 50 Jahre vergingen (Quelle: P. J. Fitzgerald, R. J. Koch, Delayed radionecrosis of the larynx, American Journal of Otolaryngology, 20 (4), 7-8/1999, Elsevier, New York, USA 1999). Die Deutsche Hirntumorhilfe gibt die durchschnittliche Latenzzeit mit 14 Monaten an (Quelle: Strahlennekrose als Nebenwirkung der Strahlentherapie, Deutsche Hirntumorhilfe e. V., Leipzig, https://www.hirntumorhilfe.de).
Hinweise auf Affektstörungen als Bestrahlungsnebenwirkung findet man dagegen nur sporadisch. Bezüglich des Auftreten einer Depression wird im Onko Internetportal auf Folgendes hingewiesen: „Depressive Symptome sind im Zusammenhang mit Krebserkrankungen nicht selten. Sie treten beispielsweise während der Diagnose auf, wenn der Patient erkennt, dass er wirklich Krebs hat, nach dem Abschluss der Erstbehandlung und dem Bewusstwerden, was geschehen ist, nach Operationen, die das Bild vom eigenen Körper verändern können, wenn Lebenspläne verloren gehen, wenn die Krebserkrankung trotz Therapien weiter voranschreitet. Manchmal sind die Symptome einer Depression auch die Nebenwirkung der Krebstherapie, etwa nach Bestrahlungen im Kopfbereich oder bei bestimmten chemotherapeutischen Behandlungen. Aber: Nicht jede Niedergeschlagenheit oder Mutlosigkeit ist gleich ein Anzeichen für eine Depression.“ (Quelle: Angst und Depression, Onko Internetportal der Deutschen Krebsgesellschaft e. V., Berlin, http://www.krebsgesellschaft.de/...).
Da mit der Diagnose und Therapie von Tumorerkankungen häufig mehrere potentielle Gefahrenschwerpunkte für den Patienten einhergehen, ist der Anteil der Strahlenschäden an Affektstörungen von anderen Schadenquellen abzugrenzen:
- Hirnschäden durch den Tumor selbst
- Hirnschäden nach Tumor-OPs
- Schädigungen durch die Einnahme starker Medikamente
- Schädigungen durch Chemotherapien
- Psychosoziale Stressbelastung
- Depressive Verstimmungen
Affektstörungen nach Strahlentherapie aus kausaltheoretischer Sicht
Trotz aller Technik und Vorsichtsmaßnahmen ist bei jeder Bestrahlung immer auch gesundes Hirngewebe bedroht, insbesondere bei der prophylaktischen Komplettbestrahlung des Gehirns. In Übereinstimmung mit den kausaltheoretischen Aussagen müssten Affektstörungen oder deren Verstärkungen als Folgen einer Bestrahlung mit ionisierenden Strahlen immer in Betracht gezogen werden, wenn die affektrelevanten Hirnregionen betroffen sind (→ Abbildung 42).
ABBILDUNG 42: STRAHLUNGSGEFÄHRDETE ZNS-AREALE HINSICHTLICH AFFEKTSTÖRUNGEN
Abbildung 42: Die in den verschiedenen Grautönen markierten Hirnbereiche stehen mit der Steuerung und Regulierung von Affekten im Zusammenhang und gehören im Hinblick auf die Gefahr der Entstehung oder Intensivierung einer Depression, Manie oder Bipolaren Störung zu den bestrahlungskritischen Bereichen.
Empirie (1): Allgemeine Auswirkungen ionisierender Strahlen auf das Zentralnervensystem
Die Toxizität ionisierender Strahlung ‑ insbesondere deren verheerende Wirkungen bei Bestrahlungen des kompletten Gehirns ‑ ist Fakt. Die meist aggressiven Hirntumore lassen sich jedoch in der Regel auf keine andere Weise oder nur im Verbund mit strahlentherapeutischen Verfahren bekämpfen.
Die Gefahr bleibender Schäden steigt dabei mit sinkendem Alter: Kinder und Jugendliche sind von Strahlenschäden noch stärker betroffen als Erwachsene. Das verwundert nicht angesichts des Umstandes, dass das Gehirn noch bis zum 25. Lebensjahr verschiedene Entwicklungsphasen durchläuft, und sich in dieser Zeit die Folgen sämtlicher Schädigungen ‑ egal, ob durch Strahlen, psychosozialen Stress oder eine andere Noxe ‑ erheblich potenzieren (→ Abschnitt 4.12.4, Abbildung 40).
In einer Studieninformation über die Untersuchung radiotherapeutischer Behandlungsmöglichkeiten der akuten lymphoblastischen Leukämie (ALL), durchgeführt am Universitätsklinikum Schleswig-Holstein in Kiel, wird auf kurz- und langfristige Folgen einer Schädelbestrahlung hingewiesen.
Bezüglich kurzfristiger Folgen heißt es: „Wie fast alle medizinischen Behandlungen kann auch eine Strahlentherapie Nebenwirkungen machen. Die folgenden Nebenwirkungen können bei der Schädelbestrahlung mit 12 Gy oder 18 Gy auftreten: ‑ ein sogenannter 'Strahlenkater', das heißt Kopfschmerzen, Übelkeit, Erbrechen, Appetitlosigkeit und Abgeschlagenheit. Diese Symptome treten meist nur während der ersten Behandlungstage auf; ‑ vorübergehender Haarausfall; ‑ Konzentrationsstörungen und Störungen der Merkfähigkeit, evtl. verbunden mit einem Nachlassen der schulischen Leistungen. Diese Beschwerden treten manchmal erst Wochen bis Monate nach Abschluss der Strahlentherapie auf. (...) Die (...) Nebenwirkungen können in der Regel gut behandelt werden oder bessern sich von selbst nach Ende der Strahlentherapie.“ (Quelle: wie unten).
Bezüglich langfristiger Folgen heißt es: „Es können aber auch dauerhafte Spätfolgen durch die Schädelbestrahlung auftreten: ‑ verminderte Hormonbildung im Zwischenhirn und in der Hirnanhangdrüse (Hypophyse), die möglicherweise Auswirkungen auf Wachstum und Sexualität haben können, sich aber durch Hormoneinnahmen ausgleichen lassen; ‑ leichte Beeinträchtigungen von intellektuellen Leistungen wie Konzentration und Aufmerksamkeit, Merk- und Lernfähigkeit, vor allem bei sehr jungen Kindern; ‑ Patienten mit ALL und Chemotherapie entwickeln in ca. 1 % der Fälle eine weitere Krebserkrankung (Leukämien, Tumore). Nach einer Schädelbestrahlung ist dieses Risiko nochmal höher, insbesondere für die Entwicklung eines Hirntumors. Im Zeitraum von 15 Jahren ab Diagnose der ALL muss bei ungefähr 3% der Patienten mit Schädelbestrahlung mit dem Auftreten eines Hirntumors gerechnet werden.“ (Quelle: Patienteninformation zur Schädelbestrahlung im Rahmen der Behandlung in der Therapiestudie AIEOP-BFM ALL 2009, Version 1.3, 7.1.2013, http://www.bfm‑international.org/aieop/...).
Auf diese Problematik verweist ebenfalls der Onkologe Christoph Requadt in seiner wissenschaftlichen Arbeit und bezieht sich auf eine andere Studie: „Die deutsche LESS-Studiengruppe (Late effects surveillance system) der Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH) zeigte in einer retrospektiven, multizentrischen Studie, dass die ZNS-Bestrahlung der Hauptfaktor für das Auftreten neurokognitiver Spätfolgen nach Behandlung einer ALL im Kindesalter ist.“ (Quelle: C. Requadt, Neurokognitive Spätfolgen bei Kindern und Jugendlichen mit akuter lymphoblastischer Leukämie und Hirntumor, Inaugural-Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2010, https://opus4.kobv.de/...).
Der Leiter der oben erwähnten LESS-Studiengruppe, Thorsten Langer, äußert sich über die Situation Betroffener in einem Beitrag des Berliner Tagesspiegels, in dem auch Ergebnisse einer US-amerikanischen Studie zitiert werden: „Michael musste kurz nach seinem fünften Geburtstag wegen eines Hirntumors ins Krankenhaus. Drei Mal wurde er operiert, das Gehirn und die Wirbelsäule bestrahlt (...). Das Überleben hatte einen Preis. Während die meisten kleinen Leukämiepatienten später ein normales Leben führen, ziehen Hirntumoren und ihre Behandlung oft schwere und vielschichtige Spätfolgen nach sich: Schmerzen, körperliche Schäden, auch das Denken ist beeinträchtigt. Michael etwa kann sich schlecht konzentrieren, wird schnell müde. «Wenn er sich etwas merken soll, dann geht das nur mit anschaulichen Erzählungen. Oder wenn er selbst etwas sehen und ausprobieren kann», erzählt seine Mutter. Er ist kein Einzelfall. «Wir sehen das oft in Tests», sagt Thorsten Langer, der das Late Effects Surveillance System (LESS) an der Uniklinik Erlangen leitet. «Die Kinder brauchen entweder länger als andere für eine Aufgabe oder sie geben auf, weil sie müde geworden sind. Sie sind nicht so aufmerksam.» Neues lernen, Erinnerungen abspeichern, Informationen schnell verarbeiten ‑ all das ist bei vielen von ihnen in Mitleidenschaft gezogen. An welchem Teil der Behandlung das liegt, war bisher unklar. (...) Eine Studie von zwei Krebsmedizinern von der Universität von Kalifornien in Irvine, Vipan Kumar Parihar und Charles Limoli, weist nun auf einen möglichen Mechanismus hin. Wie sie im Fachblatt 'PNAS' schreiben, verändert sich bei Mäusen je nach Stärke der Bestrahlung die Struktur wichtiger Teile des Gehirns. Die Nervenzellen im Hippokampus ‑ dem Tor zur Erinnerung ‑ hatten einen Monat nach der Behandlung 20 bis 35 Prozent weniger Dendriten gebildet, jene Fortsätze, mit denen Nervenzellen Reize aufnehmen. Außerdem waren die Verzweigungen 40 bis 70 Prozent weniger dicht. Zusätzlich änderte sich die Chemie an den Synapsen, den Verbindungsstellen zwischen den Nervenzellen. Verzweigungen, die sich gerade erst gebildet hatten, reagierten noch empfindlicher auf die Bestrahlung als andere. «Diese Veränderungen tragen wahrscheinlich zu den kognitiven Komplikationen der Patienten bei oder sind ursächlich», schreiben die Ärzte. Denn im gesunden Gehirn werden solche Verzweigungen je nach Bedarf immer wieder umgebaut und Verbindungen neu geknüpft. Ist diese Flexibilität eingeschränkt, wird es schwierig, auf Neues zu reagieren. «Das heißt nicht, dass man die Bestrahlung verteufeln sollte», sagt Langer. «Wir brauchen sie fast immer, um die Kinder zu retten.» Ärzte seien aber in der Pflicht, möglichst schonend zu behandeln und die ehemaligen Patienten mit den Folgen der Therapie nicht allein zu lassen.“ (Quelle: Jana Schlütter, Spätfolgen nach Krebs ‑ Strahlentherapie schädigt Nervenzellen, Tagesspiegel vom 16.7.2013, Berlin 2013, https://www.tagesspiegel.de/wissen/spaetfolgen‑nach‑krebs‑...).
Christoph Requadt zitiert in seiner oben erwähnten Arbeit weitere Untersuchungen zu diesem Thema: „Im Besonderen möchte ich hier auf die neurokognitiven Spätfolgen der ZNS-Prophylaxe in Form von funktionellen Veränderungen eingehen. Die ZNS-Prophylaxe besteht entweder in einer Bestrahlung des Schädels, in der intrathekalen oder systemischen Gabe von Zytostatika (z. B. Methotrexat) oder in einer Kombination dieser Therapieansätze. Durch den Einsatz einer ZNS-Prophylaxe i. R. der jeweiligen Therapieoptimierungsstudie (TOS) konnte die Häufigkeit eines ZNS-Rezidivs drastisch gesenkt werden. Diese aggressiven multimodalen Therapieansätze beinhalten ein breites Spektrum an akuten Toxizitäten und Spätschäden, denen vor den insgesamt sehr guten Therapiemöglichkeiten mit einem Event free survival (EFS) von ca. 70% eine immer größer werdende Bedeutung zukommt. Bei den Überlebenden werden morphologische Veränderungen des Gehirns in Form von mineralisierenden Mikroangiopathien (morphologisch sichtbar als Verkalkungen), Leukenzephalopathien (Nekrosen der weißen Substanz im Gehirn) und Demyelinisierungen bzw. zerebrale Atrophien in 30 ‑ 50% nachgewiesen. Berichtet wird auch über neurologisch-funktionelle Teilleistungsstörungen (Feinmotorik und Koordination) und neuropsychologische Auffälligkeiten (Merkfähigkeits- und Konzentrationsstörungen, IQ-Abfälle sowie Verhaltensauffälligkeiten und Schulprobleme) (Langer et al., 2002), die durch die Chemotherapie sowie die prophylaktische ZNS-Bestrahlung verursacht werden können. (Peylan‑Ramu et al., 1978; Brouwers et al., 1984, 1985; Bleyer und Griffin, 1980; Price et al., 1978; Williams und Davies, 1986; Ochs et al., 1983; Hertzberg et al., 1997). Dabei wird der ZNS-Bestrahlung die primär schädigende Wirkung zugeschrieben, die Kombination mit systemischer und intrathekaler Chemotherapie scheint diesen Effekt noch zu verstärken (Price und Birdwell, 1978; McIntosh et al., 1976).“ (Quelle: C. Requadt, Neurokognitive Spätfolgen bei Kindern und Jugendlichen mit akuter lymphoblastischer Leukämie und Hirntumor, Inaugural-Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2010).
Dass sich ionisierende Strahlen auch kurzfristig auf die Hirnleistungsfähigkeit negativ auswirken, wies der Radioonkologe Frederik Wenz mit seinem Team in einer am Universitätsklinikum Mannheim durchgeführten Untersuchung an erwachsenen Tumorpatienten nach. In einer Pressemitteilung aus dem Jahre 2008 anlässlich der Veröffentlichung der Studienergebnisse teilte er mit: „In einer mehrjährigen Studie, die von der Dietmar Hopp Stiftung an der Universitätsstrahlenklinik Mannheim gefördert wurde, sollte gründlich untersucht werden, ob und gegebenenfalls welche Einschränkungen der Hirnleistung durch Bestrahlung ausgelöst werden können. (...) die in den Lehrbüchern weit verbreitete Meinung, dass Nebenwirkungen neuropsychologischer Prägung erst nach mehreren Jahren auftreten, muss revidiert werden: Unsere Arbeiten zeigen erstmals, dass Beeinträchtigungen der Hirnleistungsfähigkeit in Form von Störungen des Wortgedächtnisses zwar selten sind, jedoch bereits unmittelbar während der Strahlentherapie auftreten können.“ (Quellen: Frederik Wenz, Chemo- und Strahlentherapie-assoziierte Auswirkungen auf die Hirnleistungsfähgikeit erwachsener Tumorpatienten, Pressemitteilung der Dietmar Hopp Stifung, http://dietmar-hopp-stiftung.de/..., mit Bezug auf die Studien G. Welzel, F. Wenz et al., Memory Function Before and After Whole Brain Radiotherapy in Patients with/without Brain Metastases, International Journal of Radiation Oncologiy, 72 (5), 12/2008, Elsevier, New York, USA 2008 und Wenz, Welzel et al., Acute Neurocognitive Impairment During Cranial Radiation Therapy in Patients with Intracranial Tumors, Strahlentherapie und Onkologie, 12/2008, 184 (12), Springer Berlin Heidelberg 2008).
Diese Erkenntnisse sind nicht neu, wie die oben von C. Requadt zitierten Studien zeigen. Neu ist jedoch die Bewertung als seltenes und unbedeutendes Phänomen, das vorwiegend vorgeschädigte Gehirne betrifft. Damit vertritt Wenz eine Außenseitermeinung und widerspricht auch dem jahrzehntelangen und weltweiten erfahrungsmedizinischen Wissen. In derselben Pressemitteilung heißt es nämlich weiter: „Die Forschergruppe um den Leiter der Klinik für Strahlentherapie Professor Frederik Wenz und der Diplom-Psychologin Grit Welzel konnte nun eindeutig zeigen, dass die meisten Teilfunktionen der Hirnleistungsfähigkeit durch eine Bestrahlung keinerlei Beeinträchtigungen zeigen. Vor allem bei der vorbeugenden, sogenannten prophylaktischen Bestrahlung des Gehirns konnten nur minimale und vorübergehende Einschränkungen im verbalen Gedächtnis gefunden werden. Im Gegensatz hierzu waren die Verschlechterungen der Leistungsfähigkeit bei Patienten mit Tochtergeschwülsten im Gehirn, den sogenannten Hirnmetastasen, oder mit einem Hirnödem vorübergehend etwas stärker ausgeprägt und betrafen auch das Arbeitsgedächtnis. Das vorgeschädigte Gehirn verträgt also die Bestrahlung schlechter als das gesunde Gehirn.“ (Quelle: wie oben).
Die von Wenz und Welzel im International Journal of Radiation Oncology im Dezember 2008 veröffentlichte Studie umfasste einen Zeitraum von acht Wochen. Die angegebenen Störungen des verbalen Gedächtnisses betrafen sowohl die Gruppe der Patienten mit als auch ohne Hirnmetastasen, jedoch zeigten sich die Nebenwirkungen bei der Gruppe mit Metastasen schon unmittelbar nach Beginn der Bestrahlungen. Langfristige Folgen, die über den Zeitraum von acht Wochen hinausgehen, waren nicht Teil der Untersuchung. Die zweite in der Zeitschrift Strahlentherapie und Onkologie veröffentlichten Studie kam zu vergleichbaren Schlussfolgerungen.
Es muss stark bezweifelt werden, aufgrund der Ergebnisse dieser beiden Studien auf die relative Unbedenklichkeit einer medizinischen Strahlenexpostion des Zentralnervensystems schließen zu können. Aussagen über langfristige Folgen sind auf Grundlage dieser Untersuchungen darüber hinaus überhaupt nicht möglich.
In einer neueren Studie, die am US-amerikanischen The University of Texas MD Anderson Cancer Center unter Führung des Radioonkologen Paul Brown durchgeführt wurde, untersuchte man Nutzen und Risiko einer Ganzhirnbestrahlung von 213 Patienten mit bis zu drei kleinere Hirnmetastasen. Es gab zwei Gruppen: Eine Gruppe wurde nur mit der präzisen stereotaktischen Bestrahlungsmethode (SB) behandelt, die andere Gruppe erhielt zusätzlich eine Bestrahlung des gesamten Gehirns zwecks Senkung des Rezidivrisikos. Die Patienten beider Gruppen erhielten keine zusätzliche Chemotherapie, so dass die Auswirkungen der Bestrahlungen gut zu erfassen waren. Die Studienergebnisse wurden Ende Mai 2015 präsentiert (Quellen: Risks of whole brain radiation therapy added to radiosurgery outweigh benefits for patients with limited brain metastases, MD Anderson News Release 05/31/2015, The University of Texas MD Anderson Cancer Center, Houston/USA 2015, http://www.mdanderson.org/newsroom/..., und in deutscher Sprache Keine Ganzhirnbestrahlung bei neu diagnostizierten Hirnmetastasen?, Medical Tribune vom 24.6.2015, http://www.medical‑tribune.de/...).
Schon drei Monate nach Bestrahlungsbeginn zeigten sich bedeutende Unterschiede zwischen beiden Gruppen. Der kognitive Leistungsabfall (Kurzzeitgedächtnis, Abrufzeit der Gedächtnisinhalte, verbale Artikulation) betraf 63,5% der SB-Patienten, jedoch schon 91,7% der Patienten aus der Gruppe der zusätzlich Bestrahlten. Nach sieben Monaten lag die Quote schon bei 77,8% zu 97,9%. Die ganzhirnbestrahlten Patienten beklagten darüber hinaus die wesentlich massivere Verschlecherung der Lebensqualität im Vergleich mit der anderen Gruppe. Die kognitiven Einschränkungen blieben auch bei den Langzeitüberlebenden (> ein Jahr) erhalten.
Interessant sind auch die Folgen für den weiteren Krankheitsverlauf und die Überlebensperspektive. Zwar konnte durch die zusätzliche Bestrahlung eine bessere Kontrolle der Metastasierung im Gehirn erfolgen, jedoch stiegen damit nicht die Überlebenschancen der Betroffenen. Im Gegenteil - diese hatten statistisch sogar eine kürzere Lebenserwartung.
Empirie (2): Auswirkungen ionisierender Strahlen auf Affekte und Affektstörungen
Dass es einen Zusammenhang zwischen medizinischen Bestrahlungen des Gehirns und Fällen von Depression gibt, ist allgemeiner Konsens und beruht auch auf erfahrungsmedizinischen Kenntnissen. Im schon erwähnten Informationsportal der Deutschen Krebsgesellschaft e. V. wird darauf hingewiesen (Quelle: Angst und Depression, Onko Internetportal der Deutschen Krebsgesellschaft e. V., Berlin, http://www.krebsgesellschaft.de/...).
Leider sind die Zusammenhänge zwischen medizinischen Hirnbestrahlungen und Affektstörungen wenig erforscht. Im Vergleich etwas besser ist die Datenlage über die Folgen von radioaktiven Verstrahlungen der Umwelt, denn hier wurde eine Zunahme psychischer Erkrankungen festgestellt, sei es aufgrund der Reaktorunglücke in der Ukraine (Tschernobyl) bzw. Japan (Fukushima) oder aufgrund der Erfahrungen nach den Atombombenexplosionen in Japan gegen Ende des zweiten Weltkriegs.
Da man sich in Untersuchungen nach solchen Katastrophen erfahrungsgemäß im Schwerpunkt mit Veränderungen des Krebsrisikos beschäftigte, ist die Datenlage bezüglich psychischer Erkrankungen aber auch hier eher unbefriedigend. Darauf verwiesen auch die Autoren einer Meta-Studie der Gesellschaft für Strahlenschutz und der deutschen Sektion des IPPNW aus dem Jahre 2006, die sich mit den gesundheitlichen Folgen nach der Tschernobyl-Katastrophe auseinandersetzten: „Obwohl es seit einigen Jahren Daten von den Opfern von Hiroshima und Nagasaki gibt, die diesen Bereich betreffen, werden die Nicht-Krebs-Erkrankungen nur widerstrebend mit Strahlenvorfällen in Beziehung gebracht. Untersuchungen in diesem Bereich sind zusätzlich erschwert durch fehlende Daten.“ (Quelle: S. Pflugbeil, H. Paulitz, A. Claußen, I. Schmitz-Feuerhake, Gesundheitliche Folgen von Tschernobyl - 20 Jahre nach der Reaktorkatastrophe, IPPNW + Gesellschaft für Strahlenschutz, Berlin 2006, https://www.ippnw.de/commonFiles/...).
Die Schwierigkeit, Ursachen für das Auftreten von Affektstörungen nach Be- oder Verstrahlungen herauszufinden, hat aber noch mehr ‑ und tiefer liegende ‑ Gründe als die in der Meta-Studie geschilderten:
- Es gibt in der Medizin bisher kein allgemein akzeptiertes integriertes Modell, anhand dessen man direkte und indirekte Zusammenhänge zwischen ionisierenden Strahlen und Affektstörungen bzw. anderen psychiatrischen Erkrankungen erklären kann.
- Menschen mit einer Indikation für medizinische Bestrahlungen vereinen in der Regel mehrere Stressfaktoren in sich, so dass die Bewertung ihrer jeweiligen Anteile an der Genese einer Affektstörung schwierig bis unmöglich ist. Weitere infrage kommenden Noxen sind Medikamente, Chemotherapien, sonstige körperliche Folgen der Krebserkrankung und psychosozialer Stress.
- Auch die Strahlenopfer nach einer Umweltkatastrophe vereinen jeweils mehrere Stressfaktoren in sich. Es kommt häufig strahlenbedingt zu somatischen Erkrankungen und vor allem zu psychosozialem Stress.
Der erstgenannte Grund trifft in dem hier zur Diskussion stehenden (multi‑)kausalen Modell nicht zu, sind dessen Erklärungen der Mechanismen endogener und exogener Einflüsse auf das Zentralnervensystem plausibel. Was die beiden anderen Gründe betrifft, könnten nur entsprechend konzipierte Studien mit belastbaren Ergebnissen weiterhelfen.
Einen Vergleich zwischen dem Auftreten neurologisch-psychiatrischer Erkrankungen nach der Nuklearkatastrophe von Tschernobyl vom 26. April 1986 liefert die oben erwähnte Arbeit der Gesellschaft für Strahlenschutz. In der von dem Unfall betroffenen Region der nördlichen Ukraine wurde die Population Jugendlicher und Erwachsener zwischen 1987 und 1992 untersucht. Bei Erkrankungen des Nervensystems und psychischen Störungen wurden erhebliche Steigerungen festgestellt (→ Tabelle 22). Leider liegen keine Informationen über die Charakteristik und genaue Verteilung der Erkrankungen vor.
TABELLE 22: DYNAMIK NEUROLOGISCH-PSYCHIATRISCHER ERKRANKUNGEN NACH TSCHERNOBYL
Erkrankung/Organ |
1987 | 1988 | 1989 | 1990 | 1991 | 1992 |
|---|---|---|---|---|---|---|
| Psychische Störungen | 249 |
438 |
576 |
1.157 |
5.769 |
13.145 |
| Nervensystem | 2.641 |
2.423 |
3.559 |
5.634 |
15.518 |
15.101 |
Tabelle 22: Die Zahlen stammen aus der Meta-Studie der Gesellschaft für Strahlenschutz und sind einer Studie von A. Nyagu et al. entnommen. Sie zeigen die massiven Anstiege der Erkrankungen auf jeweils 100.000 Einwohner (Quelle: A. I. Nyagu et al., Medizinische Folgen der Tschnerobyl-Havarie in der Ukraine, Tschernobyl-Ministerium der Ukraine, Wissenschaftliches Zentrum für Strahlenmedizin, Akademie der Medizinischen Wissenschaften der Ukraine, Wissenschaftlich-Industrielle Vereinigung Pripjat, Wissenschaftlich-Technisches Zentrum, Kiev 1994).
Die Meta-Studie der Gesellschaft für Strahlenschutz bietet interessante Zusammenfassungen aus verschiedenen Untersuchungen, die sich mit den Folgen radioaktiver Verstrahlung auf das Nervensystem und die Psyche bzw. Affekte auseinandersetzen und auch über die Tschernobyl-Havarie hinausgehen. Aufgrunddessen sollen sie hier in ihrer ganzen Länge zitiert werden: „Die psychischen Störungen, unter denen viele ehemalige Bewohner des Gebietes um Tschernobyl leiden, können auf Schädigungen von Nervenzellen durch radioaktive Strahlung zurückzuführen sein. Diese Ansicht vertrat bereits 1992 Nadejda Gulaya vom Pallaguin Institut für Biochemie in Kiew. Wissenschaftler aus verschiedenen Ländern vertreten die These, dass die Wirkung der Tschernobyl-Katastrophe auf die geistige Gesundheit der Bevölkerung das größte Problem darstellt. (...) Die Expertengruppe Gesundheit des 'Tschernobyl‑Forums' von WHO und IAEA hat Streßsymptome, Auswirkungen auf das sich entwickelnde Gehirn kleiner Kinder, organische Gehirnschäden bei hoch strahlenbelasteten Katastrophenhelfern und Selbstmorde als die vier Bereiche ihrer besonderen Aufmerksamkeit bezeichnet. K. Loganovsky weist auf die hohe Rate an Schizophrenie-Kranken bereits unter den japanischen Atombombenüberlebenden hin, nämlich 6 Prozent. Zweifellos hätten auch die Liquidatoren von Tschernobyl das größte Risiko, an neuropsychiatrischen Störungen zu erkranken, sowohl wegen der Strahlenbelastung, als auch aus anderen Gründen nach dem Unglück. Loganovsky wies auf verschiedene Untersuchungen zum Erkrankungsrisiko für Liquidatoren an anderen als Krebserkrankungen hin, die statistisch signifikante Ergebnisse erbrachten. Demnach beträgt die Risikoerhöhung pro Gray absorbierter Dosis (excess relative risk ERR/Gy) für geistige Störungen: ERR/Gy = 0,4 (95% CI = 0,17 - 0,64); für neurologische und Empfindungsstörungen ERR/Gy = 0,35 (95% CI = 0,19 - 0,52); für hormonelle (endokrine) Störungen ERR/Gy = 0,58 (95% CI = 0,3 - 0,87) (Biriukov et al. 2001 und Buzunov et al. 2001, 2003). Unter den geistigen Störungen weisen demnach (Biriukov et al. 2001) neurotische Störungen die höchste Risikoerhöhung mit ERR/Gy = 0,82 (95% CI = 0,32 - 1,32) auf. Die höchste Risikoerhöhung überhaupt aber findet sich für Durchblutungsstörungen des Gehirns (cerebrovaskuläre Störungen) mit ERR/Gy = 1,17 (95% CI = 0,45 - 1,88) (Ivanov et al. 2000). Und neuerdings wurden für cerebrovaskuläre Störungen signifikante Risikoerhöhungen bei externen Strahlendosen größer als 150 Milligray (mGy) mit einer Risikoerhöhung ERR pro 100 mGy pro Tag = 2,17 (95% CI 0,64 - 3,69) angegeben (Ivanov et al. 2005). Allerdings würden diese Ergebnisse nicht mit Hilfe ordentlich konzipierter psychiatrischer Studien und standardisierter diagnostischer Verfahren ermittelt, sondern lediglich die Angaben des staatlichen Gesundheitssystems über geistige Störungen ausgewertet. Das Lehrbuchwissen der Psychiatrie in den Nachfolgeländern der Sowjetunion förderten jedoch eine dramatische Unterschätzung geistiger Störungen und eine Mißdeutung als physische Erkrankungen sowie falsche Diagnosen innerhalb des Systems der geistigen Störungen (etwa neurotisch anstatt psychotisch oder organisch). So habe das Gesundheitsministerium der Ukraine das Vorkommen geistiger Störungen in der ukrainischen Bevölkerung im Jahre 1990 mit 2,27 Prozent angegeben, 1995 ebenfalls mit 2,27 Prozent und im Jahre 2000 mit 2,43 Prozent. Die World Mental Health (WMH) Survey Initiative der Weltgesundheitsorganisation habe jedoch mit Hilfe standardisierter Verfahren für die Ukraine 20,5 Prozent (95% CI = 17,7 - 23,3%) ermittelt – das staatliche Gesundheitssystem unterschätze offenbar geistige Störungen um ein Zehnfaches und mehr. Das WMH-System schließt sogenannte psychologische Störungen ein wie Angst, Depression, psychosomatische Störungen, Alkoholmißbrauch, und vermeidet die Verwendung von Begriffen wie Psychose, organisch bedingte Geistesstörungen und geistige Unterentwicklung (Retardation). Eine weitere Untersuchung im Rahmen der Französisch-Deutschen Tschernobyl-Initiative mit Hilfe von standardisierten strukturierten psychiatrischen Interviews (Romanenko et al. 2004) ergab eine Verbreitung geistiger Störungen von 36 Prozent unter Liquidatoren und von 20,5 Prozent in der gesamten ukrainischen Bevölkerung. Geradezu dramatisch stellt sich die Häufigkeitsverteilung von Depressionen dar: 24,5 Prozent unter Liquidatoren und 9,1 Prozent unter der Allgemeinbevölkerung in der Ukraine (Demyttenaere et al. 2004). Eine fortschreitende Zunahme von neuropsychiatrischen Störungen wird auch unter Liquidatoren beobachtet, die von 1986 bis 1987 und besonders unter denjenigen, die 3 bis 5 Jahre lang in der Sperrzone um Tschernobyl arbeiteten. Die Häufigkeit neuropsychiatrischer Störungen unter dem Personal, das seit 1986 - 1987 dort arbeitete und Strahlendosen von mehr als 250 Millisievert (mSv) erhielt, wird mit 80,5 Prozent angegeben und für Strahlendosen unterhalb von 250 mSv 21,4 Prozent (p<0,001) (Nyagu et al. 2004). Seit 1990, so berichtet Loganovsky, wird eine Zunahme der Schizophrenie-Erkrankungen festgestellt: 5,4 pro 10.000 unter dem Personal gegenüber 1,1 pro 10.000 in der Allgemeinbevölkerung. Im Vergleich zur ukrainischen Allgemeinbevölkerung stieg die Häufigkeit von Schizophrenie unter den in der Tschernobyl-Zone arbeitenden und lebenden Menschen auf das 2,4-fache im Zeitraum 1986 - 1997 und auf das 3,4-fache im Zeitraum 1990 - 1997 an (Loganovsky & Loganovskaya, 2000). Einen unter Liquidatoren ebenfalls besonders häufig anzutreffenden Symptomenkomplex ist das Chronic Fatigue Syndrom (CFS). Für 26 Prozent der Menschen mit einer Strahlenbelastung von weniger als 0,3 Sievert treffen laut Loganovsky (2000, 2003) die diagnostischen Kriterien von CFS zu. Die Häufigkeit von CFS habe von 65,5 Prozent in 1990 - 1995 auf 10,5 Prozent in 1995 - 2001 abgenommen und gleichzeitig habe ein sogenanntes Metabolisches Syndrom X (MSX) von 15 auf 48,2 Prozent der Liquidatoren zugenommen. CFS und MSX werden als Ausdruck für andere neuropsychiatrische und physisch krankhafte Entwicklungen betrachtet. CFS wird auch als umweltbeeinflußte Anfälligkeit und Anzeichen für eine sich anbahnende Neurodegeneration, für kognitive Beeinträchtigungen und neuropsychiatrische Störungen gesehen. Die linke Hirnhälfte scheine anfälliger zu sein als die rechte. P. Flor‑Henry berichtet, die beobachteten depressiven Zustandsbilder und klinischen Syndrome wie Schizophrenie und CFS, die bei einem hohen Prozentsatz der Liquidatoren anzutreffen sind, gingen einher mit hirnorganischen Veränderungen vor allem der linken Großhirnhemisphäre (bei Rechtshändern) was mit Hilfe des Elektroenzephalogramms (EEG) objektivierbar sei. Die Symptome äußerten sich auch in Form des Phänomens der frühzeitigen Alterung. Diese neurologischen Krankheitsbilder träten umso früher und schwerer auf, je jünger die Betroffenen zum Zeitpunkt der Strahlenbelastung waren. Ähnliche klinische Syndrome, die von EEG-Veränderungen der linken Hirnhälfte begleitet sind, werden auch bei den Liquidatoren beobachtet, die an einem akuten Strahlensyndrom gelitten haben, berichtet Flor‑Henry. Er sei überrascht, dass weder diese psychiatrischen Krankheiten noch die EEG-Veränderungen bei den russischen Veteranen des verlorenen Afghanistan-Krieges auftreten. Immerhin seien auch diese Soldaten enormem Streß ausgesetzt und wurden in ihrer Heimat - ganz anders als die Tschernobyl-Liquidatoren - nicht als Helden gefeiert. Mit Hilfe von Magnetresonanz-Tomographie (MRT), EEG und Positron-Emissions-Tomographie (PET) lasse sich jedoch der Nachweis führen, dass die Hirnveränderungen bei Tschernobyl-Liquidatoren und bei Veteranen des ersten Golfkrieges und des Bosnien-Krieges sehr ähnlich sind. Flor‑Henry führt das zurück auf die Verwendung von uranhaltigen Geschossen (depleted Uranium, DU) im Golf- und Bosnien-Krieg, die beim Aufschlag fein verteiltes Uran-238-Oxid in die Luft freisetzten, was eingeatmet werden konnte. Er habe festgestellt, dass die Opfer, die gegenüber Uran-238 exponiert waren, ähnliche neuropsychiatrische Syndrome entwickeln wie die Überlebenden der Atombombenabwürfe in Japan 1945.“ (Quelle: S. Pflugbeil, H. Paulitz, A. Claußen, I. Schmitz-Feuerhake, Gesundheitliche Folgen von Tschernobyl - 20 Jahre nach der Reaktorkatastrophe, IPPNW + Gesellschaft für Strahlenschutz, Berlin 2006, https://www.ippnw.de/commonFiles/...).
Fazit: Ionisierende Strahlung und Affektstörungen
Die Einflüsse einer medizinischen Gehirnbestrahlung auf Affekte und Affektstörungen sind noch wenig erforscht. Besser sieht es beim Verständnis der negativen Auswirkungen auf kognitive Leistungen aus, beispielsweise auf die Konzentrations‑, Merk‑ oder Lernfähigkeit. Hier wurden klare Zusammenhänge und auch langfristige Schäden nachgewiesen.
Das vermehrte Auftreten psychiatrischer Erkrankungen in nach atomaren Unfällen oder Kriegshandlungen radioaktiv verstrahlten Gebieten spricht für einen starken Kausalzusammenhang zwischen neurodegenerativ wirkender ionisierender Strahlung und Affektstörungen.
Negative Auswirkungen ionisierender Strahlungen auf Affekte und deren Beteiligung an der Entstehung bzw. Verstärkung einer Affektstörung sind auch anhand der kausaltheoretischen Modelle plausibel. Danach steigt die Wahrscheinlichkeit des Auftretens affektiver Erkrankungen durch Be- oder Verstrahlung affektrelevanter Hirnareale mit ionisierender Strahlung gleich welcher Art.
4.13.4 Kopftraumata durch Erschütterungen ▲
Im Zusammenhang mit geschlossenen Kopftraumata bzw. inneren Kopfverletzungen sind im Wesentlichen drei Erkrankungen zu unterscheiden:
- Leichtes Schädel-Hirn-Trauma ersten Grades (Gehirnerschütterung),
- Schädel-Hirn-Trauma zweiten und dritten Grades und die
- Chronisch-traumatische Enzephalopathie (CTE).
Jede massive Erschütterung des Gehirns kann ein Schädel‑Hirn‑Trauma (SHT) zur Folge haben. Durch Gewalteinwirkung von außen kommt es zu mechanisch bedingten Schädigungen des Hirngewebes aufgrund von Quetschungen, einer Hirnschwellung oder Hirnblutungen. Auf der Mikroebene sind Nervenzellen gefährdet durch eine schnelle Folge von Beschleunigung und Entschleunigung, was axonale Schäden zur Folge haben kann.
Die Gehirnerschütterung stellt dabei einen Sonderfall dar, sie wird auch als leichtes Schädel‑Hirn‑Trauma bezeichnet. Zur Abgrenzung dient die Symptomatik im ersten SHT‑Stadium, dem Akutstadium (→ Abschnitt Die Stadien des Schädel‑Hirn‑Traumas), wobei die Gehirnerschütterung durch eine kurze Zeit der Bewusstlosigkeit und vergleichsweise leichte Symptome charakterisiert ist.
Es gibt Angaben über bis zu 400 Schädel-Hirn-Verletzte auf 100.000 Einwohner bei steigender Tendenz in Deutschland (Quelle: Volker Faust, Psychosoziale Gesundheit - Kopf-Unfall und seelische Folgen, http://www.psychosoziale-gesundheit.net/seele/...).
Die Chronisch-traumatische Enzephalopathie, auch Dementia pugilistica genannt, ist dagegen eine degenerative Hirnerkrankung, die auf viele kleine Kopfstöße bzw. Gehirnerschütterungen zurückzuführen ist und seit Ende der 1920er Jahre zunächst bei Berufsboxern diagnostiziert wurde. Über die genauen hirnorganischen Folgen der Erschütterungen und Ursachen der Erkrankung herrscht bei Medizinern und Hirnforschern weitgehend Uneinigkeit. In einem kurzen Fachbeitrag aus dem Jahre 2012 beschreiben Andrew Gardner und Peter Stanwell den Stand der Diskussion (Quelle: A. Gardner, P. Stanwell, Chronisch traumatische Enzephalopathie (CTE) als Folge von Kopfverletzungen, medicalsports network 4/2012, Verlag Succida AG, Darmstadt 2012, https://www.researchgate.net/...). Über die Verbreitung der CTE gibt es keine Angaben.
Alle drei Erkrankungen betreffen nicht nur Berufsboxer, mehrere Sportarten gelten als besonders gefährlich, vor allem für die Entstehung einer Chronisch-traumatischen Enzephalopathie:
- American Football,
- Boxen,
- Eishockey,
- Rugby,
- Fußball (insbesondere durch die Annahme von Kopfbällen) und
- Wrestling.
Auch nach militärischen Kampfeinsätzen wurden häufiger geschlossene Kopfverletzungen bei Soldaten festgestellt. Bei alpinem Skifahren, Snowboardfahren, Handball, Baseball oder in der Leichtathletik kommt es vereinzelt zu Unfällen mit Schädel-Hirn-Verletzungen.
Verkehrsunfälle enden häufig mit einem Schädel-Hirn-Trauma. Alkoholiker und ältere Menschen gehören aufgrund häufiger Stürze ebenfalls zum gefährdeten Personenkreis.
Zahlreiche Informationen für Mediziner, Betroffene und Angehörige von Opfern liefert die Web‑Seite der Hannelore‑Kohl‑Stiftung (Schütz Deinen Kopf! Gehirnerschütterungen im Sport, https://www.schuetzdeinenkopf.de).
Hirngewebeschäden physikalisch-mechanischen Ursprungs entstehen auch von innen infolge eines gesteigerten Hirndrucks durch einen raumfordernden Hirntumor, eine Spontanblutung oder den Hydrocephalus genannten Liquorüberschuss. Sie sind Folgen anderer Primärerkrankungen und werden deshalb erst im Abschnitt über biologisch-medizinische Noxen erörtert.
Offene Kopfverletzungen bergen ebenfalls ein hohes Risiko, werden aus systematischen Gründen hier aber nicht thematisiert.
Die Stadien des Schädel-Hirn-Traumas
Das Schädel-Hirn-Trauma umfasst drei Phasen, wobei innerhalb der ersten Phase - dem Akutstadium - drei Schweregrade differenziert werden, mit deren Hilfe eine Gehirnerschütterung vom eigentlichen Schädel-Hirn-Trauma 2. oder 3. Grades abgegrenzt werden kann:
- Phase: Akutstadium mit Bewusstseinsstörung
Im Akutstadium entscheidet sich die Stärke des SHT, die von der Qualität der Bewusstseinsstörung und ihrer Zeitdauer abhängt. Der Schweregrad wird mit der GCS-Skala ermittelt, man unterscheidet ein leichtes (1. Grades, Gehirnerschütterung), mittelschweres (2. Grades) und schweres (3. Grades) Schädel-Hirn-Trauma.
Ein SHT 1. Grades wird auch als Gehirnerschütterung oder Commotio cerebri bezeichnet. Sie ist u. a. gekennzeichnet durch eine vorübergehende Funktionsstörung des Gehirns mit einer Bewusstlosigkeit von wenigen Sekunden bis zu zehn Minuten. Bewusstseinslücken ausschließlich hinsichtlich des auslösenden Ereignisses sind typisch für eine Gehirnerschütterung.
Ein SHT 2. Grades wird diagnostiziert, wenn die Bewusstlosigkeit zwischen zehn Minuten und einer Stunde andauert.
Bei einem SHT 3. Grades dauert die Phase der Bewusstlosigkeit länger als eine Stunde, die durch weitere Verletzungsmerkmale des Gehirns bedingt sind: Quetschungen und Druckanstieg aufgrund von Ödemen oder Blutungen. Oft sind hier chirurgische Notmaßnahmen erforderlich, um eine Druckentlastung zu erreichen.
Während eine Gehirnerschütterung mit dem Akutstadium als abgeschlossen gilt, schließen sich sowohl beim SHT 2. als auch 3. Grades jeweils eine zweite und dritte Phase an. Erst ab Schweregrad 2 liegt das eigentliche Schädel-Hirn-Trauma vor.
- Phase: Erholungs- bzw. Remissionsstadium des SHT 2. oder 3. Grades
Der Beginn der Symptomerückbildung kennzeichnet den Übergang in die Erholungsphase, die von kurzer Dauer sein kann, manchmal aber auch mehrere Jahre dauert. Es ist ein teilweises oder vollständiges Verschwinden der Symptomatik möglich.
Bleibt eine Restsymptomatik, geht das SHT in die dritte Phase über.
- Phase: SHT-Dauerschaden
Besonders bei schweren Hirnverletzungen ist eine völlige Remission oftmals nicht zu erreichen und es bleiben funktionale Handicaps. Dabei gehören Störungen des Gedächtnisses, des Erlebens und Affektstörungen zu den häufig auftretenden Dauerschäden. Auch schwere Körperbehinderungen oder ein Apallisches Syndrom (Dauerkoma) können das Ergebnis eines SHT sein.
Die Chronisch-traumatische Enzephalopathie (CTE) - eine rätselhafte Erkrankung?
Beim Schädel-Hirn-Trauma (SHT) stehen meist singuläre Ereignisse, wie schwere Stürze oder heftige Schläge auf den Kopf, mit besonders starker Wirkung und einer akuten Bewusstseinseintrübung im Vordergrund.
Gerade in Gefahrensportarten kommt es aber häufig zu zahlreichen Standardsituationen oder wenig beachteten Zwischenfällen, die sich summieren und dann zu einer Gefahr werden, beispielsweise die häufige Annahme von Kopfbällen beim Fußballspiel oder ständige ‑ auch kleinere ‑ Kopfstöße beim Boxen oder American Football. So sind US-amerikanische Profi-Footballspieler pro Jahr im Durchschnitt 950 Kopftreffern ausgesetzt, wie eine Untersuchung über den Zeitraum von sechs Jahren an der University of North Carolina at Chapel Hill in den USA ergab (Quelle: Kopfschutz: Helm auf!, National Geographic 2/2011).
Diese ständigen Erschütterungen stehen im Verdacht, mittel- bis langfristig die degenerative Hirnerkrankung CTE zu verursachen, bei der es relativ spät und im Laufe der Jahre zu immer mehr und immer stärkeren neurologisch-psychiatrischen Symptomen bzw. Erkrankungen kommt, unter anderem Depression mit Antriebsverlust, aber auch Morbus Parkinson, Demenz oder schwere Persönlichkeitsveränderungen.
Die exakten physiologischen Vorgänge, die zur CTE führen, sind noch nicht bekannt. Es gibt Annahmen über axonale Schäden, die später zur Waller-Degeneration führen, Veränderungen der Mikrogliazellen oder vaskuläre Schädigungen kleiner und kleinster Blutgefäße. Mit großer Wahrscheinlich handelt es sich um einen multifaktoriellen Prozess, bei dem alle diese vermuteten Ursachen - und ggf. noch weitere - zusammengenommen zu den hirnorganischen Schäden führen.
Schädel-Hirn-Trauma und Chronisch-traumatische Enzephalopathie aus kausaltheoretischer Sicht
Zusammenhänge zwischen Gehirnerschütterungen bzw. ‑verletzungen und Affektstörungen lassen sich anhand der kausaltheoretischen Modelle klar ableiten: Denn ob es sich um schädliche Teilchenstrahlung, Röntgenstrahlung oder von außen einwirkende Erschütterungen geht - immer müssen vor allem die affektrelevanten Hirnareale geschädigt sein, damit in der Folge eine Depression, Manie oder Bipolare Störung auftreten kann.
Die folgende Abbildung 43 stimmt daher mit der vorangehenden Abbildung 42 weitgehend überein.
ABBILDUNG 43: TRAUMAGEFÄHRDETE AREALE UND AFFEKTSTÖRUNGEN
Abbildung 43: Alle in den verschiedenen Grautönen markierten Hirnbereiche dienen der Steuerung und Regulierung von Emotionen und Affekten (→ Kapitel 1). Hier haben Traumata zwangsläufig erhebliche Auswirkungen und führen durch degenerative Gewebeschädigungen potentiell zur Entstehung einer Depression, Manie oder Bipolaren Störung.
Es gibt wenige Untersuchungen oder Studien, die eine exakte Eingrenzung hirnorganischer Ursachen von Affektstörungen nach einer Gehirnerschütterung, einem SHT oder aufgrund der CTE ermöglichen, so dass sich die Betroffenheit der in Abbildung 43 hervorgehobenen Bereiche vor allem kausaltheoretisch begründen lässt.
Zu unterscheiden von hirnorganisch bedingten Affektstörungen sind depressive Verstimmungen, die aufgrund der besonders schwierigen Lebenssituation nach einem Schädel-Hirn-Trauma oder aufgrund einer CTE zusätzlich auftreten können und charakteristisch für einen psychosozialen Stressfaktor sind (→ Abschnitt 4.12).
Der Psychiater Volker Faust zählt auf, zu welchen affektiven Problemen es aufgrund eines hirnorganischen Dauerschadens durch ein Schädel-Hirn-Trauma kommen kann (Quelle: Volker Faust, Psychosoziale Gesundheit ‑ Kopf‑Unfall und seelische Folgen, http://www.psychosoziale‑gesundheit.net/seele/...). Diese sind: Veränderungen der Grundstimmung mit Depression und/oder Manie verschiedener Schweregrade, vermehrte Erregbarkeit, Angsterkrankungen, Phobien oder Antriebsstörungen.
Gut dokumentiert sind Schäden von Hypothalamus und Hypophyse nach einem Schädel-Hirn-Trauma mit der Folge affektiver Erkrankungen (Quelle: A. Kopczak, G. K. Stalla, Schädel-Hirn-Trauma und dessen Folgen für das Hormonsystem/Stand Dezember 2012, Netzwerk Hypophysen- und Nebennierenerkrankungen e. V., Fürth 2012, https://www.glandula‑online.de/...).
Bei einem Frontalhirnsyndrom handelt es sich um einen relativ unklar definierten Fachbegriff im Falle von Schädigungen des präfrontalen Cortex, unter anderem auch nach einem SHT, die mit Veränderungen des Verhaltens und der Emotionalität einhergehen, letztere immer in Folge von Verletzungen des orbitofrontalen Cortex. Es kann zu verschiedenen affektiven Störungen kommen, beispielsweise Depression, Verlust von Interessen und Antrieb, sozialer Rückzug, Burn-out, aber auch Hyperaktivität, Manie oder Euphorie. Durch die Vernetzung des orbifrontalen Cortex mit den tiefer liegenden emotionsverarbeitenden Arealen ist eine genaue Lokalisation der Schäden oft nicht möglich, denn ‑ wie schon bekannt ‑ führen insbesondere Läsionen Letzterer zu vergleichbaren Symptomen.
Der auf Neurotraumatologie und Rehabilitationswissenschaft spezialisierte Neurologe Claus‑Werner Wallesch fasste die Erkenntnisse über das Auftreten einer Depression bzw. von Affektstörungen nach einem SHT 2009 in einem Fachartikel zusammen (Quelle: Claus-Werner Wallesch, Depression bei organischen Hirnkrankheiten, NeuroTransmitter 10/2009, herausgegeben vom Berufsverband Deutscher Nervenärzte e. V., Springer Medizin Verlag GmbH, Berlin 2009, http://www.root.webdestination.de/...): „Die Erstversorgung [Anm.: des SHT] schließt häufig, vor allem bei leichten Fällen (Gehirnerschütterung), keine nervenfachärztliche Untersuchung ein. Dies ist bedenklich, weil die unterschiedlichen Schädigungsmechanismen bei Hirntraumen in der Akutphase nicht zwingend zu offensichtlichen Symptomen wie Bewusstseinsstörungen oder Lähmungen führen. Erholt man sich von den Hirnverletzungsfolgen, spielen die individuelle und soziale Kompensationsfähigkeit und das Profil der häufig schwer zu erkennenden neuropsychologischen Verletzungsfolgen eine entscheidende Rolle. Nach schwerem SHT empfinden Betroffene Defizite der emotional-affektiven Kontrolle als am meisten belastend (Kersel et al., 2001). Auch bei leichteren SHT haben ältere Menschen ein erhöhtes Risiko, eine Depression zu entwickeln (Levin et al., 2005). Bei schweren, rehabilitationspflichtigen Hirnverletzungen nehmen Depressivität und Angst im Verlauf der stationären Rehabilitation zunächst ab. Im ersten halben Jahr nach Rehabilitation steigen Depressivität und Vitalitätsmangel dann jedoch wieder, obwohl in diesem Zeitraum die körperliche Funktionsfähigkeit weiter zunimmt. Diese Zunahme ist vermutlich durch psychosoziale Belastungen infolge alltagsrelevanter Behinderungen bedingt (...). In einer britischen Studie wurden 164 Patienten mit einem Hirntrauma ein Jahr nach Verletzung untersucht (Deb et al., 1999). Davon waren 33% schwer oder mäßig behindert. Depressionen (14%) und Panikstörungen (9%) traten signifikant häufiger als in der Normalbevölkerung auf. Fortbestehende Behinderung, Minderbegabung und psychiatrische Vorerkrankungen erhöhten das Risiko psychiatrischer Störungen. Andere Autoren geben bis zu einem Drittel behandlungsbedürftiger Depressionen im ersten Jahr nach SHT an. Bei 50% der Betroffenen setzt die Depression erst drei bis sechs Monate nach Trauma ein (Jorge et al., 2004); das Suizidrisiko Betroffener ist erhöht.“
Hirnorganische Ursachen für nach einem SHT auftretende Affektstörungen hält Wallesch für wahrscheinlich: „Es spricht einiges dafür, dass auch die Depression nach SHT eine neurobiologische, unmittelbar organische Ursache hat. US-amerikanische Psychiater fanden bei ihren depressiven Patienten nach SHT im Vergleich zu solchen ohne Depression keinen höheren klinischen Schweregrad der Hirnverletzung, jedoch ausgeprägtere Veränderungen der grauen Substanz im Stirnhirn und vermehrt Defizite in neuropsychologischen Tests von Stirnhirnfunktionen (Jorge et al., 2004). Charakteristische Symptome der Depression nach SHT sind Erschöpfbarkeit, Ablenkbarkeit, Konzentrationsstörung und psychomotorische Verlangsamung. Sie entsprechen weitgehend den neuropsychologischen Folgen eines SHT, sodass die depressive Grundlage der Beeinträchtigungen nur schwer zu erkennen ist. Gleichzeitig werden Hirnverletzungsfolgen durch die Depression verstärkt. Im Zweifelsfall sollte ein Behandlungsversuch mit Antidepressiva und begleitender Psychotherapie erfolgen. Bei einer Depression nach SHT besteht meistens gleichzeitig auch eine Angststörung (Jorge et al., 2004). Anders als Deb et al. fanden Jorge et al. vor allem generalisierte Angststörungen (bei fast der Hälfte der Patienten mit Depression nach SHT). Dies entspricht auch den eigenen Erfahrungen sowohl mit Schlaganfall als auch mit Hirntraumapatienten. Die posttraumatische Belastungsstörung als psychogene Angststörung infolge einer außergewöhnlichen Belastung kann auch nach SHT mit Bewusstlosigkeit auftreten,der ist jedoch selten. Zumeist bildet sich die Symptomatik innerhalb von Monaten weitgehend zurück, eine Chronifizierung ist die Ausnahme. Zusammenfassend spricht einiges dafür, dass Depressionen und Angststörungen nach SHT eine organische Ursache haben. Die Depression wird bei Personen mit bleibenden Behinderungen durch die psychosoziale Belastung verstärkt. Die Depression ihrerseits verstärkt die Alltagsrelevanz neuropsychologischer Defizite, die infolge des SHT bestehen. Aktuell wird zunehmend untersucht, ob auch leichte SHT vom Typ der Commotio zu anhaltenden Beschwerden und Defiziten führen können. Dazu gehören neben Fatigue-Symptomatik und neuropsychologischen Defiziten auch depressive Störungen. In einer Längsschnittuntersuchung von zuvor nie depressiven Patienten mit SHT und einem Glasgow Coma Score von zu keinem Zeitpunkt unter 13 waren eine depressive Symptomatik eine Woche nach SHT, höheres Alter und intrakranielle Läsionen im CT mit einer depressiven Episode innerhalb der nächsten drei Monate assoziiert (Levin et al., 2005). Eine kürzlich publizierte Studie mit funktioneller Kernspintomografie ergab bei Abwesenheit struktureller Läsionen Hinweise für Funktionsstörungen frontaler kortiko-subkortikaler Schleifensysteme und eine Reduktion grauer Substanz in diesen Systemen bei Patienten mit Depression ein halbes Jahr nach Commotio (Chen et al., 2008).“ (Quelle: wie oben).
Die Aussagen von Wallesch lassen den Schluss zu, dass eine strikte Unterscheidung zwischen Gehirnerschütterung und Schädel-Hirn-Trauma im engeren Sinne vor allem hinsichtlich deren potentieller Folgen nicht sinnvoll ist.
Affektstörungen bei SHT und CTE aus erfahrungsmedizinischer und empirischer Sicht
In einer relativ aktuellen kanadischen Studie unter der Leitung von Donald Redelmeier wurde festgestellt, dass das Suizidrisko nach einer Gehirnerschütterung innerhalb der Bevölkerung der kanadischen Provinz Ontario um mehr als das Dreifache höher ist (Quelle: M. Fralick, D. A. Redelmeier et al., Risk of suicide after a concussion, Department of Medicine, Sunnybrook Health Sciences Centre, University of Toronto, Ontario/Canada, Canadian Medical Association Journal, 2/2016, Ottawa/Kanada 2016, http://www.cmaj.ca/...). Bei 31 vollzogenen Suiziden pro 100.000 Einwohner und Jahr lag der Wert deutlich über dem Durchschnitt von 9,3 vollzogenen Fällen. Ereignete sich die Gehirnerschütterung an einem Wochenende, stieg die Zahl der vollzogenen Selbstmorde sogar auf 39 an, also um mehr als das Vierfache.
Das ist zwar noch kein Beleg für eine Kausalität, jedoch ein solches Szenario nach den bisherigen Feststellungen durchaus plausibel. Es könnte darüber hinaus ein Beleg dafür sein, dass die Einschätzung einer Gefahr auch leichter Schädel-Hirn-Verletzungen durch den Neurotraumatologen Claus‑Werner Wallesch nicht unrealistisch ist (→ oben).
Obwohl Pathologen schon seit den 1950er Jahren Gehirne verstorbener Sportler der Gefahrensportarten histologisch untersuchen, wird die Existenz einer CTE‑Erkrankung erst seit der Jahrhundertwende ernsthaft diskutiert. Der 2002 verstorbene American-Football-Star Mike Webster konnte mit einem Gutachten im Jahre 1999 nachweisen, dass sein massiv geistig-körperlicher Abbau auf die Zeit als National-Football-League-Profispieler (N.F.L.) zurückzuführen ist. Nach seinem Tod bestätigte die Untersuchung seines Gehirns durch den US-amerikanischen Pathologen Bennet Omalu eine Erkrankung an CTE.
Hinweise liefern vor allem die Suizide der letzten Jahre. Alle unter CTE-Verdacht stehenden N.F.L.-Profisportler begangen hauptsächlich aufgrund ihrer Depression Selbstmord. Bennet Omalu veröffentlichte im Jahre 2006 eine weitere Untersuchung des Gehirns des durch Suizid verstorbenen Footballspielers Terry Long. Omalu beschrieb das Gehirn des zum Todeszeitpunkt 45-jährigen Long als das eines an fortgeschrittener Alzheimer-Demenz erkrankten Neunzigjährigen.
Der bekannte US-amerikanische Wrestler Chris Benoit tötete sich und seine Familie im Juni 2007. Bei der Obduktion seines Gehirns stellte man fest, dass es mit dem eines an fortgeschrittener Alzheimer-Demenz erkrankten Gehirns vergleichbar war und führte das auf die vielen Schläge und Gehirnerschütterungen zurück, die Benoit im Laufe seiner Karriere erleiden musste.
Doch erst seit einer massiven Steigerung der Anzahl von Selbstmorden mehrerer Spitzensportler der N.F.L. in den Jahren 2011 und 2012 beschäftigt sich die amerikanische Öffentlichkeit intensiver mit dem Thema, auch Dank der Aktivitäten von Bennet Omalu. Traurige Beispiele sind der N.F.L.Spieler Dave Duerson im Jahre 2011 und Junior Seau bzw. Ray Easterling 2012. Football ist nämlich in Amerika nicht nur ein Thema für Profisportler; in den USA spielen knapp vier Millionen Kinder, Jugendliche und Studenten diesen beliebten Nationalsport, die nun alle als gefährdet gelten. In einem Beitrag der Online-Ausgabe der The New York Times werden einige der spektakulärsten N.F.L-Fälle beschrieben (Quelle: The N.F.L.'s Tragic C.T.E. Roll Call, Beitrag vom 3. Februar 2016, The New York Times Company, New York/USA 2016, http://www.nytimes.com/...).
Im Jahre 2012 leitete die amerikanische Neuropathologin Ann McKee eine Untersuchung über den Zustand der Gehirne 85 verstorbener Personen - davon ehemalige Berufssportler und Militärangehörige -, die häufig Erschütterungen ausgesetzt waren. Als Kontrollgruppe fungierte eine Gruppe von 18 Personen ohne Hirnverletzungen bzw. ‑erschütterungen (Quelle: A. McKee et al., The spectrum of disease in chronic traumatic encephalopathy, Brain - A Journal of Neurology, 138, 12/2015, Oxford University Press, Oxford/U. K. 2015, http://brain.oxfordjournals.org/...). McKee und ihr Team diagnostizierten in 43 Fällen CTE, das entspricht einer Rate von 63%. Aber auch andere Nervenerkrankungen waren überdurchschnittlich zahlreich vertreten. Unter anderem stellte man bei 12% der untersuchten Gehirne Schädigungen der motorischen Neuronen fest, 11% hatten Anzeichen einer Alzheimer-Demenz.
Im Rahmen dieser Studie untersuchte Ann McKee auch das Gehirn des schon erwähnten N.F.L.-Footballspielers Dave Duerson, der an einer schweren Depression erkrankte. Duerson tötete sich 2011 gezielt durch einen Schuss in den Oberkörper, um eine Obduktion des Gehirns zu ermöglichen. Er gab schon vor dem Suizid dazu seine Einwilligung und stellte sein Gehirn der Boston University School of Medicine zur Verfügung. Es wurden starke Schädigungen in den Bereichen festgestellt, die für die Regulierung und Steuerung von Emotionen zuständig sind (Quelle: Désirée Karge, Erschütterte Sportlerhirne, Bild der Wissenschaft 3/2014, Verlag Konradin Medien GmbH, Leinfelden-Echterdingen 2014, http://www.wissenschaft.de/archiv/...).
Etwa 4.000 frühere Profispieler reichten daraufhin eine Sammelklage gegen die N.F.L. ein, denn der Verband hatte jahrelange die Existenz einer Gefahr geleugnet und diese Aussage teilweise mit Gefälligkeitsgutachten zu belegen versucht.
Mittlerweile steht sogar das bisher eher unverdächtige und behäbigere Baseball unter CTE-Verdacht, nachdem sich der bekannte Baseballspieler Ryan Freel 2012 das Leben nahm. Freel suizierte sich 2012 ebenfalls aufgrund einer Depression. 2013 obduzierten McKee und ihr Team sein Gehirn und diagnostizierten CTE.
Ein Artikel der Online-Ausgabe der The New York Times aus dem Jahre 2013 zeigt Bilder von Hirnschnitten der erkrankten und verstorbenen Athleten Cookie Gilchrist, Wally Hilgenberg, Louis Creekmur, Ollie Matson, Derek Boogaard, John Grimsley und Dave Duerson und unterstellt einen Zusammenhang mit CTE (Quelle: Images of Brain Injuries in Athletes, The New York Times Company, New York/USA 2013, http://www.nytimes.com/...).
Regelmäßig wird auch nach den spektakulären Suizidfällen der Jahre 2011 bis 2012 bei Profisportlern CTE diagnostiziert, beispielweise 2017 beim Ex‑Footballspieler Aaron Hernandez. Sein Gehirn wies massive Degenerationen an Frontalhirn, Hippocampus und Amygdala auf, ebenso waren die ventrikulären Hohlräume erweitert. Hernandez verbüßte eine lebenslange Freiheitsstrafe wegen Mordes und erhängte sich in seiner Gefängniszelle. Aussagen von Ann McKee lassen darauf schließen, dass vermutlich Impulskontrolle und Aggressionshemmung durch die Hirnschädigungen beeinträchtigt waren, mit emotionaler Labilität und Wutanfällen als Folgen, wodurch sich auch strafrechtliche Fragen stellen (Quelle: Löcher im Gehirn ‑ das schwere Leiden des Ex‑NFL‑Stars Aaron Hernandez, Der Stern, Verlag stern.de GmbH, Hamburg, 11/2017, https://www.stern.de/...).
Relativ aktuell ist der Fall des ehemaligen N.F.L.-Profis Philip Adams, der unter Wahnvorstellungen und mangelnder Impulskontrolle litt und 2021 in den USA sechs Menschen und anschließend sich selbst tötete. Auch sein Gehirn wurde von Ann McKee obduziert und als an CTE erkrankt diagnostiziert (Quelle: Ein Ende des Schweigens, die tageszeitung vom 15.12.2021, taz Verlags u. Vertriebs GmbH, Berlin 2021, https://taz.de/...).
Fazit: Affektstörungen nach Schädel-Hirn-Trauma und bei Chronisch-traumatischer Enzephalopathie
Die Auswirkungen einer Schädel-Hirn-Verletzung oder einer Chronisch-traumatischen Enzephalopathie auf die Affektverarbeitung im Gehirn sind im Detail noch wenig erforscht.
Aus kausaltheoretischer Sicht ist ein solcher Zusammenhang plausibel und bedarf keiner weiteren Erläuterung (→ Abbildung 43).
Insbesondere bei Kindern, Jugendlichen und jungen Erwachsenen bis etwa zum 25. Lebenjahr wirken sich sowohl das SHT als auch viele kleine Gehirnerschütterungen im Hinblick auf die CTE fatal aus, denn deren Zentralnervensystems befindet sich noch im Aufbau. Es besteht hier kein grundsätzlicher Unterschied zu anderen Noxen, beispielsweise psychosozialem Stress (→ Abschnitt 4.12.4 über die Auswirkungen des psychosozialen Stresses in Kindheit und Jugend, insbesondere Abbildung 40 über die Entwicklung des noch jungen Gehirns).
Es mehren sich aber auch die Hinweise aus der Erfahrungsmedizin und Empirie, dass es diesen unmittelbaren Zusammenhang gibt. So sind Schädigungen der Hypothalamus-Hypophysen-Achse nach einem SHT gut belegt. Auch Verletzungen cortikaler und subcortikaler Systeme wurden nach SHT bzw. bei CTE nachgewiesen, bei CTE mehrfach durch Untersuchungen der weltweit anerkannten amerikanischen CTE-Spezialisten Bennet Omalu und Ann McKee.
Darüber hinaus erfolgten die Suizide der an CTE erkrankten Sportler meist aufgrund einer Depression ‑ ein weiterer Hinweis für einen engen Zusammenhang zwischen CTE und Affektstörungen.
Auch erkrankungsbedingt auftretende psychosoziale Stressfaktoren haben das Potential, darüber hinaus zu depressiven Verstimmungen zu führen. Die schwierigen Lebenssituationen vieler Erkrankter machen eine solche Entwicklung wahrscheinlich. Derartige psychosoziale Disstressfaktoren sind aus kausaltheoretischer Sicht potentiell zusätzliche Verursacher einer klinischen Affekterkrankung bzw. in der Lage, eine schon bestehende Affekterkrankung zu verstärken (→ Abschnitt 4.12).
4.13.5 Lärm ▲
Als Lärm bezeichnet man ein unangenehmes bzw. unerwünschtes und als störend empfundenes meist lautes Geräusch.
Die Lautstärke des Lärms bzw. dessen Schalldruckpegel wird in Dezibel (dB) gemessen. Lärmempfinden und Störpotential eines Geräuschs sind jedoch individuell. Es hängt von mehreren Faktoren ab, ob jemand ein Geräusch als unangenehm empfindet. Neben der Lautstärke sind hier das Alter und die körperlich-psychische Verfassung der Person sowie die Art oder Quelle des Geräuschs von Bedeutung.
Die subjektive Lärmempfindlichkeit betrifft vor allem Geräusche von weniger als 75 dB, was dem Schalldruckpegel durchschnittlichen Verkehrslärms entspricht. Ab einer Lautstärke von ca. 75 bis 80 dB wird Schall von den meisten Menschen als störend empfunden
Dennoch gibt hier Ausnahmen, wie das nachfolgende Beispiel zeigt: Der Geräuschpegel in einem Café oder in einer Kantine beträgt ca. 60 bis 65 dB, in einem Musikklub sind 100 dB oder mehr keine Seltenheit. Während einige Menschen Kantinenlärm jedoch als Belästigung empfinden, setzen sich andere ‑ oder auch dieselben Personen ‑ wiederum gerne in Tanzklubs wesentlich lauteren Geräuschen aus, ohne sie als Lärm zu empfinden.
Die Schmerzgrenze für Lärm liegt bei ca. 125 dB.
Lärm und Lärmempfindlichkeit entstehen im Kopf
Beim Nachdenken über die Entstehung und Verarbeitung von Schall und Geräuschen fällt einem häufig zunächst das Hörorgan ein. Der wichtigste Teil des Hörens findet jedoch im Zentralnervensystem statt. Das Hörorgan hat „nur“ die Aufgabe, Schallwellen auf bestimmte Weise in neuronale Signale umzuwandeln und weiterzuleiten, die das Gehirn anschließend in einem komplexen Prozess unter verschiedenen Gesichtspunkten verarbeitet und interpretiert: Lautstärke, Frequenz, Richtungs- und Distanzbestimmung, Einordnung bekannt/unbekannt bzw. gefährlich/ungefährlich, das Erkennen emotionaler Färbungen von Stimme oder Musik oder die Entschlüsselung verschiedener Codes, beispielsweise der Sprache.
Kenntnisse über die Art und Weise der zentralnervösen Geräuschverarbeitung sind sehr beschränkt. Das Thema wird in der Hirnforschung intensiv bearbeitet.
Folgende Hirnareale sind beim Hören involviert:
- Unteres Stammhirn (Medulla oblongata) für das Auslösen von Reflexen.
- Oberes Stammhirn bzw. Mittelhirn (Mesencephalon) mit der Colliculi inferiores, wahrscheinlich für die Richtungsbestimmung und Tonhöhenerkennung.
- Oberes Zwischenhirn (Thalamus) für die Filterung akustischer Informationen und deren Weiterleitung an das Großhirn.
- Verschiedene Großhirnkerne, beispielsweise Mandelkern oder Hippocampus, und der Gyrus cinguli für die Gefahrenerkennung, den Vergleich mit Gedächtnisinhalten und die emotionale Verarbeitung.
- Komplexe Geräuschverarbeitung einschließlich der Bewusstwerdung und Entschlüsselung der Codes durch die auditiven Areale im Schläfenlappen (Gyrus temporalis) des Cortex. Dieser Teil des Großhirns wird auch als Hörrinde bezeichnet.
Mit Hilfe des kausaltheoretischen Ansatzes ist daher auch plausibel, warum Menschen unterschiedlich lärmempfindlich sind. Die Begründung gleicht derjenigen, die das Vorhandensein unterschiedlicher Resilienzen erklärt (→ Abschnitt 1.5). Aus dieser Sicht gibt der Zustand des emotionalen Systems die Grenze der Lärmempfindlichkeit vor oder ist daran zumindesten wesentlich beteiligt, wobei hier ohne Zweifel auch das Hörorgan einen wesentlichen Anteil hat.
Außerdem können auch nicht pathologische Faktoren, beispielsweise individuelle Persönlichkeitsmerkmale, die Lärmempfindlichkeit beeinflussen.
Lärm und Gesundheit
Auch bei den negativen Auswirkungen des Lärms auf die Gesundheit denkt man meist an das Hörorgan, obwohl Lärm mittlerweile als Auslöser vieler anderer körperlicher und nervlicher Erkrankungen verdächtigt wird.
Laut einer WHO-Studie aus dem Jahre 2011 stellt Lärm das zweitgrößte Gesundheitsrisiko dar und erhöht die Zahl von Herz-Kreislauf-Erkrankungen, kognitiven Störungen, Tinnituserkrankungen und Schlafstörungen sowie die Stressbelastung mit der Konsequenz des Verlusts von bis zu 1,6 Millionen gesunden Lebensjahren in der Bevölkerung der Europäischen Union pro Jahr (Quelle: Burden of disease from environmental noise, World Health Organization Regional Office Europe, Kopenhagen/Dänemark 2011, http://www.euro.who.int/...).
Lärm wirkt sowohl akut als auch chronisch:
- Akute Auswirkungen des Lärms sind Schreck- und Stressreaktionen und Beeinträchtigungen des Schlafs.
- Chronische Auswirkungen des Lärms resultieren zusätzlich aus den durch langfristigen Stress ausgelösten psycho-physiologischen Veränderungen und betreffen die in der WHO-Studie genannten Erkrankungen.
Die genauen Zusammenhänge zwischen Lärm und Affektstörungen sind unklar, jedoch gibt es verschiedene Hypothesen.
Nächtlicher Lärm als Auslöser von Affektstörungen?
Schlafprobleme werden häufig als Begleiterscheinungen oder Symptome affektiver Erkrankungen aufgefasst. Eine weitere weit verbreitete Vermutung besteht darin, Schlafstörungen als Folgen anderer Symptome einer Affektstörung zu betrachten.
Beruhen die Schlafprobleme eindeutig auf Lärm und ist die betroffene Person auch an einer Affektstörung erkrankt, drängt sich die wesentlich plausiblere Vermutung eines kausalen Zusammenhang zwischen nächtlichem Lärm und Symptomen affektiver Störungen auf: Könnten also die ständige nächtliche Geräuschverarbeitung bzw. lärmbedingte Schlafstörungen nicht auch die Ursachen bestimmter Symptome affektiver Erkrankungen sein? Und wie ist eine solche Kausalkette zu begründen?
Nächtlicher Lärm behindert gleich auf mehrere Weisen den Schlaf:
- Einschlafstörungen,
- Durchschlafstörungen,
- Wiedereinschlafstörungen und
- frühes Erwachen.
Die psychische Gesundheit könnte durch nächtlichen Lärm auf zwei verschiedene Weisen gefährdet sein:
- Alle Unterbrechungen bzw. Störungen des Nachtschlafs belasten Körper und Gehirn direkt, denn sie führen zu einem Erholungsdefizit. Die unmittelbaren Folgen ‑ Müdigkeit und Unausgeglichenheit am Tage ‑ haben langfristig das Potential, mindestens zu depressiven Verstimmungen führen oder eine bestehende Affekterkrankung zu verstärken.
- Ein zweites Problem ist die unbewusste Informationsverarbeitung von Geräuschen, die auch in der Nacht stattfindet. Selbst wenn es nicht zu einem Erwachen kommt - die zusätzliche Tätigkeit und Ablenkung des Gehirns während der Nacht verursacht überflüssigen Disstress, denn Geräusche des Nachts werden vom Gehirn als Gefahr interpretiert und beeinträchtigen dadurch die wichtigen Tiefschlafphasen. Darüber hinaus steigen dadurch bei Tage Müdigkeit und Unausgeglichenheit sowie die Reizbarkeit, während die Leistungsfähigkeit sinkt. Peter Lercher, Sozialmediziner der Medizinischen Universität Innsbruck, weist auf die physiologischen Hintergründe dieser Entwicklung hin: „«Die Ursache dafür liegt im Ansteigen der Stresshormone im Blut», erklärt Peter Lercher von der Medizinischen Universität Innsbruck. «Selbst wenn die Betroffenen den Lärm nicht bewusst wahrnehmen [d. h. im Schlaf, Anmerkung des Verfassers], steigt der Puls um durchschnittlich drei bis fünf Schläge pro Minute.»“ (Quelle: Paul Klammer, Kathrin Schwarze‑Reiter, Stresshormone im Blut: Unerwartete Geräusche wertet das Gehirn als Alarmsignal, Focus Magazin 31/2013, Focus Magazin Verlag, München 2013).
Verschiedene physiologische Mechanismen der Stressbewältigung bei nächtlichem Lärm, zu denen hauptsächlich das Ausschütten von Stressshormonen zählt, fördern mittel- bis langfristig die Entstehung oder Verstärkung psychiatrischer Erkrankungen. Diese komplexen Mechanismen werden im Abschnitt über psychosozialen Disstress ausführlich behandelt, so dass sie an dieser Stelle nicht mehr erörtert werden müssen (→ Abschnitt 4.12).
Direkte physiologische Folgen ständiger Schlafprobleme werden im Zusammenhang mit Auswirkungen körperlicher Erkrankungen im Abschnitt 4.14 über biologisch-medizinische Noxen noch detailliert erörtert. Dennoch sollen die Ergebnisse einer Studie über die Folgen von Schlafstörungen schon an dieser Stelle erwähnt werden, da Zusammenhänge zwischen Lärm als Ursache von Schlafstörungen augenscheinlich sind und die Schlussfolgerungen der Wissenschaflter in dieser Studie eine alternative Erklärung für Zusammenhänge zwischen Schlafstörungen und psychiatrischen Symtpomen bietet.
Die in den USA durchgeführte Untersuchung befasste sich mit allgemeinen zentralnervösen Auswirkungen von Schlafstörungen und ‑mangel. Seung-Schik Yoo, Matthew P. Walker und ein Team der Harvard Medical School in Boston und der University of California in Berkeley „...haben sich in der 2007 veröffentlichten Untersuchung eingehend mit den Prozessen auseinandergesetzt, die infolge von Schlafmangel im Gehirn ablaufen. Sie fanden heraus, dass gestörter Schlaf die Kommunikation zwischen Mandelkern (Amygdala) und Frontallappen massiv beeinflusst und sogar unterbricht. Während der Mandelkern die Bewertung emotionaler Reize steuert, werden in einem Teil des Frontallappens, dem präfrontalen Kortex, die kognitiven Prozesse reguliert und dabei situationsgerechtes Handeln instruiert. Fehlender Schlaf kann folglich die Fähigkeiten des Gehirns einschränken, Emotionen zu verarbeiten und angemessen auf emotionale Reize zu reagieren. Walker glaubt, dass der gestörte Kommunikationsverlauf zwischen Amygdala und Frontallappen so schließlich zum Auftreten psychiatrischer Symptome führt.“ (Quelle: Stephanie Schersch, Schlafstörungen - Ursachen für psychische Krankheiten, Pharmazeutische Zeitung 35/2009, Govi-Verlag Pharmazeutischer Verlag GmbH, Eschborn 2009 bzw. Yoo, Walker et al., The human emotional brain without sleep - a prefrontal amygdala disconnect, in: Current Biology, Vol. 17, Issue 20, Elsevier, New York, USA 2007, http://www.sciencedirect.com/...).
Tageslärm als weiterer psychosozialer Disstressor
Was für nächtlichen Lärm und Geräusche gilt, spielt zweifellos auch beim Tageslärm eine Rolle. Disstress stellt hier die Hauptgefahr dar, der über vergleichbare oder identische physiologische Stressbewältigungsprozesse, wie sie vom Schlafmangel bekannt sind, die Entstehung oder Verstärkung psychiatrischer Störungen begünstigt (→ Abschnitt 4.12).
Auch der Psychiater und Neurologe Volker Faust macht in seinen Schriften zur Psychosozialen Gesundheit Stressreaktionen für die Probleme verantwortlich, die durch Lärm ausgelöst werden (Quelle: Volker Faust, Lärm ‑ Umweltproblem Nr. 1 und Geissel unserer Zeit: Seelische, körperliche und psychosoziale Folgen, Arbeitsgemeinschaft Psychosoziale Gesundheit, http://www.psychosoziale-gesundheit.net/..). Faust führt Beeinträchtigungen der geistigen Leistungsfähigkeit, Gemütsstörungen, hormonelle Fehlsteuerungen und somatische Störungen aufgrund Veränderungen verschiedener Regulationsmechanismen von Nierenfunktion, Muskulatur, Atmung oder Blutwerte auf Stressreaktionen zurück.
Empirie: Lärm und Affektstörungen
Lärm als Auslöser gesundheitlicher Probleme gewinnt als Forschungsgegenstand immer mehr an Bedeutung. Verschiedene Studien weisen Zusammenhänge zwischen Lärm und affektiven Erkrankungen nach.
Ein Langzeit-Forschungsprojekt des Zentrums für Urbane Epidemiologie (CUE) der Medizinischen Fakultät der Universität Duisburg-Essen ergab einen Zusammenhang zwischen Straßenlärm und der Erkrankung an einer Depression. Insgesamt wurden 3.300 anfänglich gesunde Probanden im Alter zwischen 45 und 75 Jahren in der Ruhrregion Essen-Bochum-Mülheim in die Untersuchung einbezogen. Die fünfjährige Studie begann im Jahre 2000. Sie ergab einen Anstieg der Fälle von Depression bei Personen, die stark von Straßenverkehrslärm betroffen waren. Das Risiko, an einer Depression zu erkranken, stieg in diesem Falle um 25% bei einer Lärmbelastung von über 55 (24‑Stunden‑Mittelwert) bzw. 50 dB (Mittelwert Nachtlärm). Menschen mit geringerem Bildungsgrad reagierten empfindlicher auf Lärm (Quellen: Traurige Beschallung, Pressemitteilung der Universität Duisburg-Essen vom 26.11.2015 und Ester Orban, Kelsey McDonald et al., Residential Road Traffic Noise and High Depressive Symptoms afte Five Years of Follow-up: Results from the Heinz Nixdorf Recall Study, 11/2015, Centre for Urban Epidemiology (CUE), Institute for Medical Informatics, Biometry and Epidemiology, Essen 2015, http://ehp.niehs.nih.gov/...).
Aus Anlass des Brenner-Basis-Tunnelbaus wurden die Konsequenzen steigenden Verkehrslärms vom Innsbrucker Sozialmediziner Peter Lercher untersucht und 2007 veröffentlicht. Er stellte einen signifikanten Anstieg der Einnahme von Schlafmedikamenten bei Personen mit gesundheitlichen Vorbelastungen in Fällen steigender Lärmbelastung fest. Ähnliches gilt sowohl für die Häufigkeit einer diagnostizierten Depression als auch einer Medikation mit Psychopharmaka, hier jedoch bei der gesamten Gruppe der Befragten (Quelle: Peter Lercher, Public Health Studie BBT, Sektion Sozialmedizin der Medizinischen Universität Innsbruck, 2007 ‑ 2014, https://www.i-med.ac.at/hygiene/bbt-files/...).
Im Jahre 2009 wurden im Auftrag des Umweltbundesamtes Gesundheitsdaten von etwa 1,02 Millionen Personen hinsichtlich der Einnahme blutdrucksenkender Medikamente und psychischer Erkrankungen rund um den Flughafen Köln-Bonn ausgewertet. Diese nach den Autoren Eberhard und Claudia Greiser benannte Greiser‑Studie stellte bezüglich der Häufigkeit psychischer Erkrankungen bei Frauen ein erhöhtes Erkrankungsrisiko fest und betraf vor allem nächtlichen Fluglärm. Im Abschlussbericht des Umweltbundesamtes wird auch auf eine Studie aus dem Jahre 2006 derselben Autoren verwiesen, die zu einem ähnlichen Ergebnis bei der Untersuchung des Arzneimittelverbrauchs der Bewohner in der Umgebung des Köln-Bonner-Flughafens gekommen ist. Bei steigender Fluglärmbelastung fanden sich bei Frauen sowohl erhöhte Verordnungshäufigkeiten als auch höhere Verordnungsmengen für Antidepressiva (Quelle: Greiser/Greiser, Risikofaktor nächtlicher Fluglärm, Schriftenreihe Umwelt & Gesundheit 01/2010, Umweltbundesamt, Dessau-Roßlau 2010, https://www.umweltbundesamt.de/...).
Unter der Leitung des Psychologen Rainer Guski startete im April 2011 die NORAH‑Studie über die Auswirkungen des Verkehrslärms in Bezug auf den Flug-, Schienen- und Straßenverkehr im Rhein-Main-Gebiet rund um den Flughafen Frankfurt/Main auf die Gesundheit und Lebensqualität. Grundlage der Untersuchung war ein stresstheoretischer Ansatz über die Folgen von Lärm. Die Analysen wurden im Jahre 2013 abgeschlossen (Quelle: Pressemitteilung über die Lärmwirkungsstudie im Auftrag der Gemeinnützigen Umwelthaus GmbH, Kelsterbach, auch: http://www.norah-studie.de).
Die umfangreichen Untersuchungen basierten auf telefonischen Befragungen, medizinischen Einzelerhebungen (Blutdruck-Monitoring, Schlafuntersuchungen, individuelle Risikofaktorenbestimmungen hinsichtlich Vorerkrankungen oder Rauchverhalten) und kinderpsychologischen Untersuchungen an bis zu 24.000 Bewohnern der Rhein-Main-Region. Ebenfalls wurden ca. 1,5 Millionen Krankenversichertendaten ausgewertet. Ergänzend wurden mehrere tausend Bewohner im Umkreis der Flughäfen Berlin, Köln/Bonn und Stuttgart befragt. Für die Untersuchungen des Lärmeinflusses auf die Gesundheit war der Arbeitsmediziner Andreas Seidler und das Institut für Arbeits- und Sozialmedizin der TU Dresden verantwortlich. Ziele der Studie waren die Ermittlung von Zusammenhängen zwischen der regionalen Lärmbelastung und dem Auftreten von Herz-Kreislauf-Erkrankungen, (Brust-)Krebs und Depression.
Hinsichtlich der Teilergebnisse und Erkenntnisse über das Depressionsrisiko in der Rhein-Main-Region soll aus dem Endbericht der Studie zitiert werden: „Schließlich lässt sich in unserer Fallkontrollstudie ein Zusammenhang zwischen allen drei Verkehrslärm-Arten (Fluglärm, Straßenverkehrslärm, Schienenverkehrslärm) und der Diagnose einer depressiven Episode feststellen. Die Risikoerhöhung pro 10 dB Pegelanstieg ist beim Fluglärm mit 8,9% zwar höher als beim Straßenverkehrslärm (4,1%) und beim Schienenverkehrslärm (3,9%). Allerdings finden sich bei höheren Fluglärm-Pegeln ebenso wie bei höheren Schienenverkehrslärm-Pegeln wieder sinkende Depressions-Risikoschätzer (im Sinne einer umgekehrten 'U'‑Form), und das lineare Modell bildet die Expositions-Risiko-Beziehung beim Fluglärm wie beim Schienenverkehrslärm nicht adäquat ab.“ (Quelle: Seidler et al., NORAH Noise-related annoyance, cognition and health ‑ Verkehrslärmwirkungen im Flughafenumfeld, Endbericht, Band 6, TU Dresden, Medizinische Fakultät und Gemeinnützige Umwelthaus GmbH, Kelsterbach vom 9.10.2015, http://www.laermstudie.de/...).
Im Gegensatz zu den Ergebnissen der Studie über die Region rund um den Köln-Bonner-Flughafen gibt es keine Anzeichen für geschlechterspezifische Auswirkungen. Im Köln-Bonner-Raum ergab die Greiser-Studie des Jahres 2009 lediglich bei Frauen einen Zusammenhang zwischen (Nacht‑)Fluglärm und psychiatrischen Erkrankungen, die Analysen im Rahmen der NORAH-Studie kamen nicht zu einem derartigen Ergebnis: „Generell finden sich in unserer Fallkontrollstudie bei getrennter Analyse von Männern und Frauen keine systematischen Unterschiede in der Höhe der Verkehrslärm-bezogenen Risikoschätzer für die vorgenannten Erkrankungen. Auch bei gesonderter Analyse für die einzelnen Krankenkassen-Arten zeigen sich keine systematischen Unterschiede hinsichtlich der Risikoschätzer. Da die einbezogenen Krankenkassen deutlich unterschiedliche Versichertenklientele aufweisen, spricht dieser Befund für die externe Validität der Ergebnisse.“ (Quelle: wie oben).
Im Endbericht kommen die Autoren bezüglich der Depression zu folgendem Schluss: „Insgesamt weisen die Ergebnisse unserer sekundärdatenbasierten Fallkontrollstudie mit vertiefender Befragung auf einen Zusammenhang zwischen einer Exposition gegenüber Verkehrslärm und der Entstehung eines Herzinfarktes, eines Schlaganfalls, einer Herzinsuffizienz sowie einer depressiven Episode hin. (...) Die höchsten mit Verkehrslärm verbundenen Erkrankungsrisiken zeigten sich in Bezug auf den 10-dB-Pegelanstieg für die Diagnose einer depressiven Episode – und zwar statistisch signifikant für alle drei Verkehrsarten. (...) Bei Straßenverkehrslärm zeigten sich die höchsten Risiko-Anstiege pro 10 dB Pegelanstieg bei depressiven Episoden (4,1%), Herzinfarkt (2,8%), Herzinsuffizienz (2,4%) und Schlaganfall (1,7%). Bei Schienenverkehrslärm betrugen die höchsten Risiko-Anstiege pro 10 dB Pegelanstieg für depressive Episoden 3,9% (allerdings durch ein lineares Modell nicht adäquat abgebildet), Herzinsuffizienz 3,1%, Herzinfarkt 2,3% und Schlaganfall 1,8%. Bei Fluglärm sind die höchsten Risiko-Anstiege pro 10 dB Pegelanstieg bei depressiven Episoden (8,9%, allerdings durch ein lineares Modell nicht adäquat abgebildet) und Herzinsuffizienz (1,6%) zu finden.“ (Quelle: wie oben).
Hinweis: Nur einen Tag nach der Veröffentlichung der NORAH-Studienergebnisse wurden diese von einigen Ärzten angezweifelt, insbesondere wegen einer angeblichen Verharmlosung der Risiken für die Entstehung von Herz-Kreislauf- und Krebs-Erkrankungen und unterstellter methodischer Fehler.
Fazit: Lärm als Verursacher Affektiver Störungen
Gleicht man Erkenntnisse über die Auswirkungen des Lärms und die Mechanismen der Lärm- und Geräuschverarbeitung mit den kausaltheoretischen Szenarien ab (→ Kapitel 1), drängt sich die Möglichkeit eines Kausalzusammenhangs zwischen Lärm und Affektstörungen schon auf den ersten Blick auf, denn viele emotionsverarbeitende Hirnregionen übernehmen auch beim Hörvorgang wichtige Funktionen.
Als Teile einer Schnittmenge von für beide Funktionen zuständigen Hirnareale sind hier Mittelhirn, Thalamus, Mandelkern, Hippocampus und Gyrus cinguli zu nennen, ggf. noch weitere, denn die Hirnforschung hat den Hörvorgang bisher nur höchst unvollkommen entschlüsselt.
Neben einer ungünstigen Wirkung nächtlichen Lärms für die Erholung des Gehirns durch Schlafstörungen sind die entscheidenden lärmbedingten Auslöser Affektiver Störungen höchstwahrscheinlich mittel- bis langfristiger Disstress und dadurch verursachte physiologische nächtliche Anpassungsreaktionen, durch die es im Gehirn aufgrund einer zu hohen und chronsichen Ausschüttung nervensystemschädigender Stresshormone zu Schädigungen von Nerven- und Gliazellen kommen kann*. Die zahlreichen schädigenden Stressanpassungsmechanismen werden in Abschnitt 4.12 ausführlich beschrieben. Die beiden Kausalketten lauten:
- Nachtlärm → Schlafprobleme → Geringe Erholung → Schädigung des ZNS → Affektstörung
- Lärm → Disstress → Disstressverarbeitung → Schädigung des ZNS → Affektstörung
*Hinweis: Ein objektiver Vergleich beider Szenarien ist ‑ zumindest zum gegenwärtigen Zeitpunkt ‑ nicht möglich. Von daher beruht die hier vorgenommene Gewichtung auf einer Einschätzung des Autors.
4.13.6 Zellatmung, reaktive Sauerstoff-Spezies (ROS) und oxidativer Stress ▲
Hoch agressive reaktive Sauerstoff-Spezies (ROS, für den englischen Fachterminus Reactive Oxygen Spezies) sind eine negative Begleiterscheinung der (endogenen) Zellatmung. Es gibt zwei Arten von ROS: freie Sauerstoffradikale und nicht radikale stabile reaktive Sauerstoffverbindungen. Freie Sauerstoffradikale sind beispielsweise das früher als Superoxid bezeichnete Hyperoxid und Hydroxyl, wichtige reaktive Sauerstoffverbindungen sind Ozon, Hydroperoxid und Wasserstoffperoxid. Da ROS immer in Verbindung mit Sauerstoff auftreten, werden sie ‑ abgeleitet vom lateinischen Begriff Oxygenium für Sauerstoff ‑ auch Oxidantien genannt.
ROS können zellschädigende Prozesse auslösen und bedrohen sämtliche Gewebearten und Moleküle einer Zelle.
ROS sind nicht ausschließlich das Ergebnis der Zellatmung. Die Einleitung zu Beginn des Hauptabschnitts 4.13 enthält eine grobe systematische Übersicht über Zellprozesse und exogene Noxen, die ROS zur Folge haben (→ Abschnitt 4.13 oben), beispielsweise ionisierende Strahlen, und die in den Abschnitten über die jeweiligen Noxen erörtert werden. Daher treffen die meisten Aussagen und Informationen zu den ROS-Folgen in diesem Abschnitt 4.13.6 auch auf diese Noxen zu.
Potentielle positive ROS-Funktionen im Zellstoffwechsel
Studien deuten drauf hin, dass Wasserstoffperoxid und Hyperoxid auch bei der Signalübertragung und der Blutgefäßerweiterung (Vasodilatation) im Gehirn eine positive Rolle spielen. Hier ist der Kenntnisstand aber noch gering (Quellen: S. G. Rhee, Redox signaling: hydrogen peroxide as intracellular messenger, 6/1999, EMM Experimental & Molecular Medicine, Nature Publishing Group, London/UK 1999) und K. T. Kishida, E. Klann, Sources and targets of reactive oxygen species in synaptic plasticity and memory, Antioxidants & Redox Signaling, 2/2007, Mary Ann Liebert, Inc., publishers, New Rochelle, New York/USA, 2007).
Etwaige positive stoffwechselrelevante Funktionen der ROS sollen jedoch kein Thema sein, hier geht es ausschließlich um deren negative Folgen als Verursacher von Zellstress durch Oxidation.
ROS als Abfallprodukte der Atmungskette
ROS als Abfallprodukte der Atmungskette sind Teil der Zellatmung, mit der die Energieversorgung einer Zelle sichergestellt wird. Die Abläufe, bei denen der weitaus größte Teil des Sauerstoffs verarbeitet wird, finden in spezialisierten Zellorganellen ‑ den Mitochondrien ‑ statt.
In der Atmungskette übertragen vier Proteinkomplexe (Komplex I, II, III und IV) in vier Stufen vier Elektronen und Wasserstoff-Atome auf den Sauerstoff. Die Ergebnisse sind Energie in Form von ATP und Wasser. Idealerweise sollte Sauerstoff dabei vollständig zu Wasser abgebaut werden, leider entkommt dem ein Teil und genau hier entstehen schädliche ROS.
Die Schätzungen über die Mengen an ROS, die in der Atemkette entstehen, sind relativ ungenau. In der Literatur wird angegeben, dass 1 bis 3%, manchmal sogar bis zu 10% des Sauerstoffs in potentiell gefährliche ROS-Moleküle umgewandelt wird, vor allem Hyperoxid, Hydroxyl und Wasserstoffperoxid.
Das antioxidative Schutzsystem und oxidativer Stress
Die Zellen sind den ROS jedoch nicht völlig hilflos ausgeliefert und verfügen über ein komplexes Schutzsystem. Etwa 80% der ROS-Schädlinge werden im Regelfalle sofort vom antioxidativen Enzym, der mitochondrialen Superoxid-Dismutase, inaktiviert. Aber auch die verbleibenden 20% ROS sind immer noch sehr gefährlich und müssen möglichst vollständig beseitigt werden. Hier kommen weitere zelleigene Enzyme und Substanzen zum Einsatz, ebenso durch Nahrung zugeführte antioxidative Mikronährstoffe, beispielsweise Vitamin A, Vitamin C, Vitamin E, Carotin, Kupfer, Selen oder sekundäre Pflanzenstoffe.
Kann eine Zelle die Menge schädlicher ROS nicht mehr vollständig neutralisieren, liegt oxidativer Stress vor, und es kann zu einer Schädigung des Zellgewebes kommen. Kein oxidativer Stress liegt vor, wenn sämtlichen ROS durch die antioxidativen Zellschutzsysteme neutralisiert werden. Das bedeutet: ROS sind nur eine hinreichende, jedoch nicht ausreichende Bedingung für oxidativen Stress. Es gehört auch noch die Unfähigkeit einer Zelle dazu, die ROS komplett zu neutralisieren. So führt auch eine erhöhte ROS-Menge nicht automatisch zu oxidativen Zellschädigungen, wenn die Zelle über besonders leistungsfähige ROS-Schutzsysteme verfügt.
Wie sich oxidativer Zellstress auf Lipide, Proteine und Aminsoäuren auswirken kann
Oxidativer Stress gefährdet Lipide und Lipoïde durch Lipidperoxidation und damit insbesondere innere und äußere Zellmembranen, die zum größten Teil aus Lipiden bestehen und bei denen freie Sauerstoffradikale Elektronenraub begehen. Mit dem Entreißen von Elektronen wird die radikalen Eigenschaften auf die Lipide/Lipoïde transferiert, so dass es zu einer Kettenreaktion und gegenseitigem Elektronenraub kommt, bis die Elektronenübertragung durch antioxidative Prozesse gestoppt wird (→ Abschnitt 4.13.3 über die molekular-atomare Wirkung ionisierender Strahlung).
Bis zu einem gewissen Grad sind Zellen in der Lage, Membrandefekte aufgrund von Lipidperoxidation nachträglich zu reparieren.
Von der Proteinoxidation sind sowohl größere Proteine als auch kleinere Peptide und sogar Aminosäuren betroffen. Auch die Proteinoxidation umfasst spezifische Veränderungen einzelner Moleküle durch Elektronenraub, aber auch eine Zerlegung von (Poly‑)Peptidketten oder Proteinverklumpung ‑ letzteres auch Aggregation genannt ‑ und die Bildung von Peptid‑Cross‑Links, bei der sich eine Vielzahl von (Poly‑)Peptiden zu einem dreidimensionalen Netz miteinander verknüpfen. Durch Zerlegung und Aggregation verlieren die Proteine ihre Zellfunktionen und können andere Zellfunktionen behindern, zum Teil werden radikale Eigenschaften auf sie übertragen.
Auch gegen pathologische Proteinveränderungen kann eine Zelle nachträglich Maßnahmen ergreifen. 80% aller schadhaften Proteine werden mit Hilfe von Chaperonen zu den Proteasomen befördert und von diesen geschreddert und entsorgt (Quelle: Christiane Richter-Landsberg, Olaf Goldbaum, Thomas Stahnke, Gehirnzellen im Stress, Einblicke Nr. 44, Herbst 2006, Carl von Ossietzky Universität Oldenburg, Oldenburg 2006, http://www.presse.uni‑oldenburg.de/einblicke/44/richter‑landsberg.pdf).
Kleinere Schäden nach Lipidperoxidation oder Proteinoxidation können demnach durch Reparaturmaßnahmen folgenlos bleiben. Oftmals entstehen jedoch mehr oder weniger starke Prozessprobleme oder es tritt sogar der Zelltod ein (→ Abschnitt Oxidativer Stress und Zelltod).
Da die Atmungskette in den Mitochondrien stattfindet und dort naturgemäß die größte Menge an ROS entstehen, sind die Membranen und Proteine dieser für die Energieversorgung wichtigen Organellen durch Lipidperoxidation und Proteinoxidation besonders gefährdet.
Mutationen von Zell-DNA und mitochondrialer DNA durch oxidativen Stress
Neben Lipidperoxidation und Proteinoxidation stellen DNA-Schäden durch oxidativen Stress eine weitere, erhebliche Bedrohung einer Zelle dar. Ausführliche Erläuterungen grundsätzlicher Ursachen und Folgen von DNA-Schäden und Mutationen sind in den Abschnitten 3.3, 4.2 und 4.4 nachzulesen.
Schäden der Zellkern-DNA werden, falls Reparaturen misslingen, zu somatischen Mutationen (→ Abschnitt 3.3) und diese werden auf alle nachfolgenden Tochterzellen übertragen. Von Mutationen sind potentiell alle Protein-/Peptid-Gene, ncRNA-Codes bzw. ganze Chromosomen betroffen.
Die sich außerhalb des Zellkerns befindenden Mitochondrien verfügen über eine eigene mitochondriale DNA (mtDNA) und unterliegen daher ebenfalls einer Mutationsgefahr. Erschwerend kommt für die Mitochondrien hinzu, dass die schädlichen Oxidationsprozesse naturgemäß in unmittelbarer Nähe zu ihnen stattfinden und deren DNA dadurch besonders gefährdet ist.
Unter dem Begriff der somatischen Mutationen werden folgende bleibenden DNA-Veränderungen verstanden:
- Basenoxidation = Elektronenraub durch reaktive Substanzen, z. B. freie Sauerstoffradikale.
- Desaminierung = Fehlerhafte Basenpaarungen aufgrund chemischer Veränderungen an der DNA.
- Depurinierung = Die Purinbasen Adenin und Guanin wurden vom DNA-Gerüst abgespalten.
- Depyrimidinierung = Wie Depurinierung, jedoch mit den Pyrimidinbasen Cytosin und Thymin.
- Brüche von Chromosomen ohne weitere Veränderungen an der Chromosomenstruktur.
- Brüche von Chromosomen mit der Folge diverser Chromosomenstrukturveränderungen.
Mögliche Konsequenzen von Mutationen reichen von keinen bis hin zu massiven Auswirkungen ‑ je nach Größe oder Position auf der DNA. Bei schwerwiegenden Mutationen kann es zum Zelltod kommen, im Idealfall durch eine geregelte Zell-Apoptose, häufig jedoch durch eine Zellnekrose mit weiteren Belastungen für umliegende Zellen (→ nachfolgenden Abschnitt Zelltod als Folge oxidativen Stresses).
Das Entarten zu einer Tumor- oder Krebszelle ist nach einer Mutation der Zell-DNA ebenfalls möglich. Krebszellen werden von einem gesunden Immunsystem zwar meist erkannt und beseitigt, manchmal dennoch übersehen und entwickeln sich zu einem Karzinom.
In den nachfolgenden Abschnitten geht es im Schwerpunkt um (somatische) Mutationen der Zell-DNA. Zweifelsfrei können auch Keimzellen bei oxidativem Stress mutieren, was jedoch den Kausalfaktor der ursprünglichen elterlichen Erbinformtion betrifft (→ Abschnitt 4.7 des Kapitels 4 A).
Zelltod als Folge oxidativen Stresses
Eine der übelsten Konsequenzen oxidativen Stresses ist der Zelltod, der nach derzeitigem Stand der Forschung auf zwei grundsätzliche Weisen erfolgen kann:
- Die Apoptose unterliegt einem streng regulierten, aktiven Prozess, bei dem die Zellbestandteile kontrolliert in Einzelbausteine zerlegt und aufgelöst werden, hauptsächlich mit Hilfe von Caspase-Enzymen. Endonukleasen „häckseln“ die DNA in Stücke, die Zelle schnürt sich in kleine Vesikel-Bläschen ab und wird von Makrophagen und Neutrophilen phagozytiert bzw. „gefressen“. Die Zelle stirbt, ohne Nachbargewebe zu schädigen. Im Gehirn sind hauptsächlich Mikrogliazellen für die Beseitigung des „Zellschrotts“ zuständig.
- Die Nekrose läuft demgegenüber unkontrolliert ab. Die Zellen schwellen an, die äußere Membran wird zerstört und die Zellsubstanzen gelangen in den extrazellulären Raum. Dies führt zu Entzündungen und infektiösen Reaktionen. Folge: Durch die Zell-Nekrose kann auch gesundes Nachbargewebe geschädigt werden.
Im Jahre 2005 beschrieben Yuan et al. einen Hybridmechanismus, der Apoptose und Nekrose miteinander vereinigt und nannten ihn Nekroptose (Primärquelle: C. Yuan, A. Degterev et al., Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury, 2005, Nature Chemical Biology 1 (2), Nature Publishing Group/USA, New York 2005 oder Sekundärquelle Thorsten Lieke, Nekroptose, Laborjournal 11/2011, LJ‑Verlag OHG, Freiburg, Freiburg 2011).
Apotose und Nekrose stehen daher wahrscheinlich nur am Anfang und Ende einer ganzen Bandbreite von Möglichkeiten eines Zelltods. Dazu schreibt der Biologe Jürgen Zitzler in seiner Dissertation: „Obwohl es während der Apoptose und der Nekrose zu unterschiedlichen morphologischen Ausprägungen kommt, gibt es Hinweise darauf, dass es sich bei diesen beiden Formen um die Extreme eines ineinander verlaufenden Spektrums von Möglichkeiten handelt, wie eine Zelle sterben kann (Leist et al. 1997). Sowohl in Geweben als auch in Zellkulturen existieren beide Formen nebeneinander. In Neuronen wurde gezeigt, dass ein und derselbe Stimulus, verabreicht in geringen Konzentrationen, zu Apoptose und in hohen Konzentrationen zur Nekrose der Zellen führen kann (Bonfoco et al.1995).“ (Quelle: Jürgen Zitzler, Oxidativer Stress-assoziierter neuronaler Zelltod und die Identifikation neuroprotektiver Gene durch ein neuartiges Screening-System, Fakultät für Biologie der Ludwig-Maximilians-Universtität München, München 2004, https://edoc.ub.uni-muenchen.de/...).
Das vermutete Phänomen mehrerer Zwischenformen wird auch durch die 1998 entdeckte Oxytose gestützt, die nicht mit der Oxydose (Sauerstoffvergiftgung) verwechselt werden darf. Jürgen Zitzler erwähnt in seiner Arbeit diese durch oxidativen Stress assoziierte Zelltodmöglichkeit ebenfalls: „Daneben kann es auch zu einer zeitlichen Aufeinanderfolge beider Formen kommen. Tan und Maher beschrieben eine neue Form des Zelltodes, die Oxytose, bei dem der durch oxidativen Stress induzierten Zelltod sowohl Charakteristika von Apoptose als auch Nekrose aufweist (Tan et al. 1998).“ (Quelle: wie oben bzw. die Originalquelle Tan, Wood, Maher, Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells, in: Journal of Neurochemistry 7/1998, Wiley-Blackwell, Hoboken, New Jersey/USA 1998, http://www.ncbi.nlm.nih.gov/...).
Zell- und Organschädigung durch oxidativen Stress der Atmungskette
Schon aufgrund der großen Sauerstoffmenge, die für die Energieversorgung verarbeitet werden muss, trägt oxidativer Stress aufgrund der Atmungskette eine Hauptverantwortung für Lipid- bzw. Proteinveränderungen und DNA-Mutationen und damit für zelluläre Alterungsprozesse, natürlich immer im Zusammenhang mit fehlendem oder suboptimalem Oxidationsschutz und ungenügenden Reparaturmechanismen.
Neben der Möglichkeiten eines Zelltodes führt die Tatsache, dass ein bestimmter Anteil mutierter Zellen ‑ nämlich solche, deren Prozessstörungen unterhalb einer Apoptose-, Nekrose- oder Oxytose-Schwelle liegen ‑ nicht beseitigt wird, im Laufe der Zeit zu einer Anhäufung suboptimal funktionierender Zellen in einem Organ.
Insbesondere kommen im Zeitablauf potentiell weitere Veränderungen und Mutationen hinzu, so dass sich die Anzahl suboptimal funktionierender Zellen nach und nach vergrößert und zu entsprechenden Organproblemen führt
Im Abschnitt 4.2 werden die Konsequenzen speziell von DNA-Mutationen für die Zellalterung ausführlich beschrieben. Am Ende des Abschnitts wird zusammengefasst, was unter einer mutationsinduzierten Zellalterung zu verstehen ist. Die beschriebenen Mechanismen spielen für das Verständnis der Folgen von ROS eine große Rolle (→ Abschnitte 4.2.1 bis 4.2.3 des Kapitels 4 A). Die Entwicklung einer steigenden Anzahl von Mutationen wird als DNA‑Fehlerakkumulation bezeichnet, wodurch der Ablauf der Proteinbiosynthese immer stärker gestört wird und von einer kontinuierlichen wachsenden Anzahl von Zellprozessproblemen auszugehen ist. Da die DNA hauptsächlich proteinsynthesesteuernde ncRNA-Codes enthält, sind vor allem ncRNA-Moleküle betroffen mit den Folgen immer massiveren Proteinbiosynthesemodulationsstörungen.
Auch Energieversorgungsprozessstörungen aufgrund von Schädigungen der Mitochondrien tragen zur Zellalterung bei.
Quintessenz: Zell- und Organalterung sind vor allem Resultate von Lipidperoxidationen, Proteinoxidationen und DNA-Mutationen, verursacht durch ROS der Atmungskette. Da die Nervenzellenerneuerung und -reparatur im Zentralnervensystem unter herkömmlichen Bedingungen nicht überall möglich, können deren Auswirkungen vor allem im Gehirn besonders fatal sein.
Anhand des 3‑Stufen‑Modells (→ Abschnitt 1.4) sind die Folgen dieser Entwicklung für das Gehirn im Detail nachzuvollziehen.
Endogen-induzierte und exogen-induzierte ROS
Die Menge an ROS aus der Atmungskette einer Zelle ist nicht konstant. Es ist davon auszugehen, dass deren Anzahl, die unter normalen Lebensbedingungen entsteht, als eine natürliche Mindestmenge zu betrachten ist. Unter bestimmten - meist negativen - Lebensumständen können ROS jedoch bedenklich ansteigen.
Die jeweilige ROS-Situation wird mit Hilfe der Begriffe endogen-induziert bzw. exogen-induziert differenziert:
- Bestimmte Mengen stoffwechselbedingter ROS lassen sich auch bei einer optimalen Lebensführung nicht vermeiden. Diese unvermeidlichen ROS werden als endogen-induziert bezeichnet.
Würde ein Organismus im Laufe seines Lebens nur durch endogen-induzierten oxidativen Stress belastet, könnte man den sich daraus ergebenden Alterungsprozess, beispielsweise durch akkumulierte DNA-Mutationen oder Zellverlust, als einen Teil des natürlichen Alterungsprozesses bezeichnen.
- Darüber hinaus bestehen zusätzliche Gefahren bei einem höheren oder sehr hohen Aufkommen von ROS. Das ist in Bezug auf die Zellatmung immer dann der Fall, wenn eine Zelle viel Energie erzeugen muss und damit naturgemäß auch mehr ROS entstehen und die Zellschutzsysteme unter Normalbedingungen zur Neutralisation dieser großen Mengen nicht mehr ausreichen.
Erhöhte ROS-Mengen und deren Schäden werden als exogen-induziert bezeichnet. In diesem Falle unterliegt der Organismus einem beschleunigten Alterungsprozess. Dies gilt vor allem dann, wenn ROS langfristig bzw. dauerhaft erhöht sind.
Hinweis: Exogen-induzierte ROS entstehen nicht nur durch eine intensivere Zellatmung, auch verschiedene exogene Noxen, beispielsweise über das natürliche Maß hinausgehende Mengen ionisierender Strahlung (→ Abschnitt 4.13.3), können zu einem ROS-Anstieg in einer Zelle führen. Auch diese Noxen und die damit verbundenen Zellschäden werden als exogen-induziert bezeichnet und führen potentiell zu einem beschleunigten Alterungsprozess.
Was die Menge atmungsbedingter ROS beeinflusst
Folgende Faktoren beeinflussen die Höhe exogen-induzierter ROS ‑ und damit oxidativen Stress ‑ im Zusammenhang mit der Zellatmung:
- der Energiebedarf eines Organs,
- die unabhängig vom Energiebedarf zugeführte Sauerstoffmenge und
- die neun Zellschwachstellen in Summe, insbesondere jedoch die ursprüngliche Erbinformation, somatische Mutationen der Zell-DNA mit Auswirkungen auf Gene und ncRNA-Codes und die Versorgung mit Antioxidantien.
Zu 1.: Verschiedene Situationen führen zu einem höheren Energiebedarf als gewöhnlich, der mit einem entsprechend höheren Sauerstoffverbrauch und zwangsläufig auch einer größeren Menge schädlicher ROS und der Gefahr oxidativen Stresses einhergeht, während ein ruhiger Stoffwechsel mit vergleichsweise geringem Energieverbrauch entsprechend weniger ROS produziert.
Betroffen davon ist der gesamte Körper, einschließlich des Gehirns. Vor allem folgende Szenarien lassen den Energieverbrauch ansteigen:
- starke geistige Beanspruchung,
- schwere körperliche Arbeit,
- intensive sportliche Leistungen,
- Erkrankungen des Körpers, zum Beispiel Schilddrüsen- oder maligne Erkrankungen,
- Erkrankungen des Zentralnervensystems und
- langfristiger psychosozialer Stress (→ Abschnitt 4.12).
Zu 2.: Auch die unabhängig von der Stoffwechsellage zugeführte Sauerstoffmenge ist für das Gehirn als Bestimmungsfaktor für exogen-induzierte ROS relevant. Sie kann aufgrund seltener ‑ meist außergewönlicher ‑ Situationen höher sein als üblich. Es gilt: Je mehr Sauerstoff dem Gehirn zugeführt wird, desto höher ist die Menge zellatmungsinduzierter ROS. Dies ist ein gutes Beispiel dafür, dass ein lebensnotwendiger Kausalfaktor, nämlich Sauerstoff, bei Überdosierung schädliche Folgen haben kann.
Eine Sauerstofftoxikose bzw. Oxydose ‑ auch Sauerstoffvergiftung ‑ entsteht bei einer zu hohen Konzentration von Sauerstoff in der Atemluft. Hier werden Zentralnervensystem, Gefäßsystem und Lungen durch ROS hoch belastet.
Eine Sauerstoffvergiftung kann zu schweren Hirnschäden bzw. zum Tode durch massenhaftes Absterben von Nervenzellen führen. Der Untergang der Zellen geschieht chaotisch und ungeregelt durch Zell-Nekrose, was einen zusätzlichen hohen Gewebeschaden zur Folge haben kann. Eine Sauerstoffvergiftung kommt zum Glück jedoch nur äußerst selten und hauptsächlich bei folgenden, eher außergewöhnlichen Situationen vor:
- Unter Wasser herrscht ein höherer Druck als auf dem Land, und dieser Druck steigt mit zunehmender Wassertiefe an. Da bei höherem Druck mehr Sauerstoff im Blut angereichert wird, besteht beim Gerätetauchen generell die Gefahr einer Sauerstofftoxikose und diese steigt, je tiefer man taucht. Daher müssen Taucher genau auf das von ihnen verwendet Gasgemisch in Abhängigkeit der Geräteart achten. Etwas Ähnliches gilt natürlich auch für Bergsteiger, die Atemgeräte verwenden.
- In der Anästhesie bzw. der Intensivmedizin ist die Sauerstoffvergiftung ein ärztlicher oder pflegerischer Kunstfehler, wenn Patienten während einer Operation oder Intensivtherapie zuviel Sauerstoff erhalten, was an einer fehlerhaften Beatmungseinstellung, an einem technischen Defekt des Beatmungssystems oder an der Unaufmerksamkeit des Arztes bzw. Pflegepersonals liegen kann. Aufgrund der intensiven Beobachtung und Kontrolle von Patienten und deren Vitalwerten sollten solche Zwischenfälle allerdings heutzutage so gut wie nicht vorkommen. In solchen medizinischen Situationen kann es ebenfalls zu einer Sauerstoffunterversorgung kommen. Der Vollständigkeit halber muss erwähnt werden, dass auch ein Sauerstoffmangel zu oxidativem Stress führen kann (→ Abschnitt 4.9).
Während schwere Sauerstoffvergiftungen immer schwere Hirnschäden zur Folge haben, die nicht unbemerkt bleiben können, werden Sauerstoffvergiftungen leichterer Art häufig zunächst nicht bemerkt oder nach dem Abklingen ihrer akuten Symptome von den Betroffenen meist schnell wieder vergessen.
Nichtsdestrotz können auch leichtere Vergiftungen chronische Schäden an Nerven- und Gliazellen affektrelevanter Hirnareale hinterlassen, die ‑ obwohl zunächst ohne psychiatrische Probleme zu verursachen ‑ zu einem beliebigen Zeitpunkt nach dem Ereignis für Affektstörungen (mit-)verantwortlich sind, beispielsweise weil weitere Negativfaktoren dazugekommen sind.
In einem solchen Falle würde ein Betroffener auch bei einer Erinnerung an das damalige Geschehen wahrscheinlich nie auf den Gedanken kommen, dass seine psychiatrische Erkrankung etwas mit einer Jahre zurückliegenden leichteren Sauerstoffvergiftung zu tun hat. Das gilt im übertragegenen Sinne für fast alle ZNS-Erkrankungen, beispielsweise Morbus Parkinson oder Demenz.
Zu 3.: Das körpereigene antioxidative Schutzsystem ist für die Synthese und den Einsatz körpereigener Schutzbarrieren gegen Oxidantien verantwortlich. Etwa 80% der ROS aus der Atmungskette werden durch zwei Formen des mitochondrialen Enzyms Superoxid-Dismutase neutralisiert. Weitere antioxidative Enzyme sind die Katalase, Peroxidasen oder Reduktasen. Ebenfalls stark antioxidant wirkende Substanzen des Körpers sind Albumin, Bilirubin, Gluthation, Harnsäure oder das Epiphysenhormon Melatonin.
Effektivität und Effizienz der antioxidativen Schutzsysteme werden ‑ wie sämtliche anderen Zellprozesse auch ‑ von neun Zellschwachstellen beeinflusst (→ Abschnitte 4.2 bis 4.10). Das heißt: Eine optimale Funktionsfähigkeit antioxidativer Prozesse setzt voraus, dass die acht Kausalfaktoren in korrekter Menge und Form zur Verfügung stehen und alle Zellen möglichst keine DNA-Mutationen aufweisen, die im Zusammenhang mit dem antioxidativen Zellschutz eine Rolle spielen könnten.
Hier haben drei Zellschwachstellen eine ganz besondere Bedeutung, nämlich...
- die ursprüngliche Erbinformation,
- die Akkumulation von DNA-Mutationen und
- antioxidant wirkende Mikronährstoffe bzw. Antioxidations-Synergisten.
Die ursprüngliche Erbinformation bestimmt die grundsätzliche Funktionsfähigkeit des körpereigenen antioxidativen Schutzsystems. Ist die Erbinformation in diesem Sinne als „gut“ zu bezeichnen, sollte der Organismus grundsätzlich in der Lage, die körpereigenen Antioxidantien in ausreichender Menge zu synthetisieren und alle notwendigen Prozesse durchzuführen. Diese Erbinformationsqualität betrifft vor allem Peptid-Gene und ncRNA-Codes, die im weitesten Sinne mit dem Aufbau antioxidativer Schutzsystemen im Zusammenhang stehen. Ob das antioxidative System eines Menschen grundsätzlich gut oder weniger gut in der Lage ist, ROS zu neutralisieren, hängt also auch von der polygenetischer Qualität der Erbanlagen ab (→ Abschnitt 4.7). In diesem Sinne gibt es Menschen, die „von Natur aus“ ein schlechteres Schutzsystem haben, weil familär und damit polygenetisch vorbelastet.
Es gibt derzeit keine Hinweise darauf, dass Mutationen einzelner DNA-Gene oder -Codes zu einer gravierenden Störung des antioxidativen Systems führen würden, die dann nämlich als Erberkrankungen einzustufen wären. Zwar sind Mutationen des Superoxid-Dismutase-Gens bekannt und stehen sogar im Verdacht, an der Genese der Amyotrophen Lateralsklerose (ALS) beteiligt zu sein, jedoch nicht aufgrund von Verlusten der anti-oxidativen Proteineigenschaften, sondern weil diese Mutationen zu einer Verklumpungsneigung der Superoxid-Dismutase und zum Zelltod führen können.
Die im Laufe der Zeit akkumulierten Schäden durch somatische Mutationen in Nerven- und Gliazellen des Gehirns und damit einhergehende, sich häufende Probleme bei der Proteinbiosynthese wirken auch auf die Prozesse des antioxidativen Schutzsystems, das im Laufe der Zeit immer mehr an Qualität einbüßt (→ Abschnitt 4.2) und oxidativen Stress ansteigen lässt.
Die ausreichende Versorgung mit Antioxidantien und Antioxidations-Synergisten aus der Nahrung ist daher zweifellos ein wichtiger externer Faktor für die Qualität antioxidativer Prozesse. Fallen vermehrt ROS an, beispielsweise bei einem permanent hohen Energieverbrauch des Gehirns aufgrund von Stress, sollten dementsprechend auch mehr Anitoxidantien zur Verfügung stehen (→ Abschnitt 4.4).
Wichtige Radikalfänger sind die Substanzen der Vitamin‑E‑Gruppe oder das natürliche Flavonol Rutin. Andere Antioxidantien wie Vitamin C bieten sich an, an Stelle wichtiger körpereigener Stoffe zu oxidieren; sie werden Reduktionsmittel genannt. Eine dritte Gruppe von Substanzen, die selber nicht antioxidant wirkenden Antioxidations-Synergisten, sind für die Regeneration verbrauchter Antioxidantien notwendig, darunter zählen beispielsweise viele Fruchtsäuren.
Fünf weitere Aspekte, welche die oxidative Stressbelastung des Gehirns beeinflussen
Speziell bei der Diskussion der Oxidations-Gefahren für das Gehirn sind noch fünf weitere Einflussfaktoren zu berücksichtigten. Diese zeigen, dass das Zentralnervensystem im Vergleich zu anderen Organgen grundsätzlich stärker durch oxidativen Stress in Mitleidenschaft gezogen wird.
So ist der Energiebedarf des Gehirns enorm. Seine Masse, die nur etwa 2% des Körpergewichts eines Erwachsenen ausmacht, verbraucht etwa die Hälfte der täglichen Glukose bei einem normalen, ausgeglichenen Stoffwechsel. In Situationen erhöhten Verbrauchs entsprechend mehr. Das Gehirn ist damit das am stärksten von oxidativem Stress betroffene Organ, denn hier wird mit einem Anteil von 20% am Gesamtenergiebedarf entsprechend viel Sauerstoff benötigt und verbraucht.
Dazu kommt der Umstand, dass das Gehirn einen hohen Fettgehalt hat, denn in den Membranen der Nerven- und Gliazellen ist eine Menge mehrfach ungesättigter Fettsäuren gebunden, die sehr oxidationsgefährdet sind. Ebenso betrifft das Lipoïde, beispielsweise Phosphatidlycholin oder andere Phospholipide. Im Gehirn spielt die Lipidperoxidation daher eine besonders negative Rolle.
Das Gehirn ist also schon unter normalen Bedingungen einem hohen oxidativen Stress ausgesetzt. Dazu kommt jedoch noch ein weiteres Problem: Das Gehirn, speziell die Nervenzellen und die Myelin-bildenden Oligodendrozyten, verfügt leider nur über ein begrenztes antioxidatives Schutzsystem, verglichen mit dem der meisten Körperzellen. Dieser Umstand wird in einem Beitrag der Pharmazeutischen Zeitung beschrieben: „So ist die Aktivität vieler antioxidativer Enzyme in Neuronen reduziert; die Katalase-Aktivität beträgt hier nur zehn Prozent der in der Leber (...). Erstaunlicherweise genügt bereits die physiologische exzitatorische Neurotransmission durch Glutamat, um in Neuronen einen hohen Grad an DNA-Schäden zu induzieren. Damit steigt die basale Aktivität des an der DNA-Reparatur beteiligten Enzyms Poly-ADP-Ribose-Polymerase (PARP) dramatisch an; sie erreicht dort das etwa 70-fache der Aktivität in Gliazellen (...). Oligodendrozyten, die die Myelinscheiden der Neurone bilden, weisen weniger als die Hälfte an Glutathion und Glutathionreduktase-Aktivität sowie nur etwa 15 Prozent der Glutathionperoxidase-Aktivität von Astrozyten auf (...). Darüber hinaus fehlen ihnen eine Mangan-Superoxiddismutase (Mn‑SOD) (...) und auch Metallothionin, ein Cystein-reiches Antioxidans und Bindungsmolekül für Zink- und Kupferionen (...).“ (Quelle: Oliver Ulrich, Tilman Grune, Freie Radikale - Angriff auf Neurone, Pharmazeutische Zeitung, 46/2001, Govi-Verlag Pharmazeutischer Verlag GmbH, Eschborn 2001).
Der Tod von Nervenzellen scheint darüber hinaus sehr häufig durch die ungünstige Zell‑Nekrose charakterisiert zu sein, wie eine Studie von Bonfoco, Krainc, Ankarkrona et al. aus dem Jahre 1995 zeigte, auf die der Biologe Jürgen Zitzler in seiner Arbeit verweist (Quelle: wie oben). Zell-Nekrosen werden häufig von zusätzlichen entzündlichen oder immunologischen Reaktionen begleitet, die gesundes Nachbargewebe schädigen.
Auch wenn die strikte Annahme über die begrenzte Teilungsfähigkeit von Nervenzellen in den letzten Jahren zurückgenommen werden musste, unterscheiden sich Neuronen in diesem Punkt immer noch massiv von Körperzellen. Es ist auf jeden Fall so, dass durch Apoptose, Nekrose oder Oxytose verschwundene Nervenzellen nicht so einfach oder im Normalfall eben nicht ersetzt werden können. Das gilt auch für durch oxidativen Stress bedrohte Oligodendrozyten.
Exkurs: Hyperventilation
Bei einer akuten Hyperventilation, also dem plötzlich zu schnellen und zu tiefen Ein- und Ausatmen, wird zuviel Kohlendioxid abgeatmet mit der Folge eines sinkenden Kohlendioxid-Partialdrucks im Blut. Zwar gelangt durch die schnelle Atmung auch vermehrt Sauerstoff in die Lungen, da die Blutsauerstoffsättigung im Normalfall aber (fast) 100% beträgt, nimmt das Blut auch durch Hyperventilation nicht mehr Sauerstoff auf.
Während des Hyperventilierens stellt die höhere Sauerstoffzufuhr demnach kein Problem dar. Die Abnahme des Kohlendioxid-Partialdrucks des Blutes bewirkt jedoch aufgrund komplexer biochemischer Reaktionen einen Anstieg des Blut-pH-Wertes, was mit verschiedenen physiologischen Veränderungen einhergeht, hauptsächlich einer Hypokalzämie und verminderem Blutkalziumspiegel.
Im Gehirn führt die niedrige Kohlendioxid-Konzentration sogar zu der paradoxen Situation, dass sich dort die Blutgefäße verengen und die Sauerstoffversorgung des Gehirns sinkt.
Die Ursachen der akuten Hyperventilation sind vielzahlig, häufig ist sie jedoch psychisch bedingt und tritt in Situationen mit starker Angst- bzw. Panikstimmung auf.
Affektstörungen durch oxidativen Stress aus kausaltheoretischer Sicht
Die Gefahr eines Ausbruchs oder der Verstärkung von Affektstörungen besteht immer dann, wenn oxidativer Stress zu Funktions- und Gewebeschäden in den für die Entstehung und Verarbeitung von Emotionen zuständigen Hirnarealen führt (→ nachfolgende Abbildung 44).
Die graphischen Darstellungen noxenbedingter Schädigungen sind - unabhängig von der jeweiligen Noxe - grundsätzlich identisch, da die kausaltheoretischen Prinzipien überall gleich sind. Daher unterscheidet sich die Abbildung 44 auch fast nicht von den vorhergehenden Abbildungen 43, 42 oder 38.
ABBILDUNG 44: OXIDATIVER STRESS IN AFFEKTRELEVANTEN HIRNAREALEN
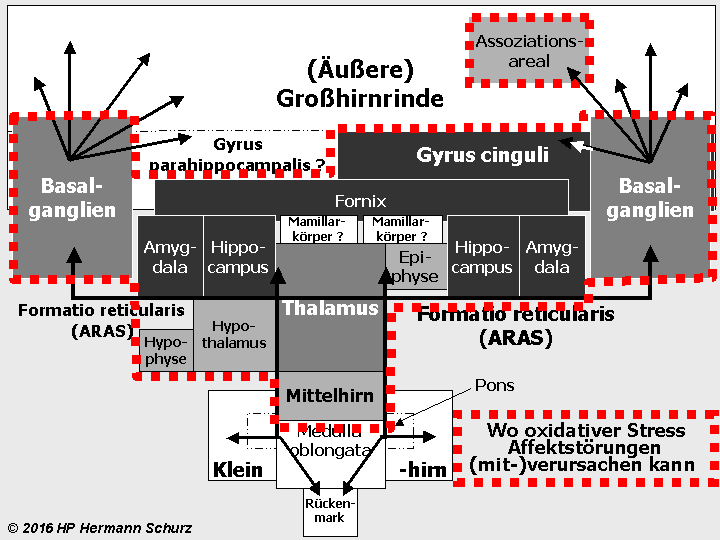
Abbildung 44: Alle in verschiedenen Grautönen markierten Areale dienen der Steuerung und Regulierung von Emotionen und Affekten. Hier hat oxidativer Stress zwangsläufig erhebliche Auswirkungen, denn oxidationsbedingte Funktions- und Gewebeschädigungen führen in diesen Arealen potentiell zur Entstehung Affektiver Störungen.
Empirie: Oxidativer Stress und die Genese von Affektstörungen
In den nachfolgend zitierten Studien hat man Zusammenhänge zwischen Affektstörungen und oxidativer Stressbelastung untersucht, meist an lebenden Patienten oder Probanden. Im Rahmen einer Post-Mortem-Studie (Tanja M. Michel et al.) dienten ausnahmsweise Hirnpräparate Verstorbener der Analyse von Merkmalen oxidativen Stresses.
In den hier beispielhaft aufgeführten Arbeiten stand immer die Gesamtbelastung mit oxidativem Stress im Fokus und demnach nicht ausschließlich oxidativer Stress der Atmungskette. Diese Unschärfe in der Analyse hat ihre Ursache in der Schwierigkeit, bei der Untersuchung eine Differenzierung nach Stressquellen durchführen zu können. Zwar gibt es ROS-Substanzen, die in besonders großer Zahl in der Atmungskette anfallen, eine ausschließliche Fokussierung auf diese Substanzen oder deren Marker hilft aber nicht wirklich weiter, da ROS auch an anderen Stellen im Zellstoffwechsel entstehen, beispielsweise nach einer Belastung durch ionisierende Strahlung, beispielsweise Röntgenstrahlen oder Radioaktivität.
Da in der Atmungskette aber der größte Teil des Sauerstoffs verstoffwechselt wird, kann man von einem hohen Anteil der endogenen Zellatmung am gesamten oxidativen Stressvolumen ausgehen. So sind die Zellatmungsprozesse ununterbrochen aktiv, während andere Quellen oxidativen Stresses, beispielsweise ionisierende Strahlung, meist nur in ganz bestimmten Fällen eine Rolle spielen. Solche Ausnahmen sind darüber hinaus meist bekannt, beispielsweise bei der Untersuchung von Strahlengeschädigten, und dementsprechend können Studien-Design und Studienauswertung entsprechend angepasst werden.
In der überwiegenden Anzahl der Untersuchungen werden bestimmte Stressparameter im Blut zum Nachweis oxidativen Stresses herangezogen. Hier eine Auswahl dreier geeigneter Substanzen:
- Carbonylproteine (CP)
Es handelt sich um Proteine, die unter ROS-Einfluss oxidieren und im Blutplasma gemessen werden. Die Werte sollten zwischen 80 und 200 pmol/mg betragen. Bei neurodegenerativen Erkrankungen wurden erhöhte Werte um die 600 pmol/mg festgestellt (Quelle: Univ.-Prof. Mag. Dr. Joachim Greilberger, Mag. Dr. Peter Moser, Seminar Oxidativer Stress, November 2011, Medizinische Universität Graz, Institut für Physiologische Chemie, http://www.ralf‑kollinger.de/wp/...).
- 8-Hydroxydesoxyguanosin (8-OHdG)
8-OHdG ist u. a. ein Marker für das besonders häufig vorkommende Sauerstoffradikal Hydroxyl. Es entsteht bei Hydroxyl-assoziierten oxidativen Veränderungen der DNA und kann im Blutserum und -plasma bestimmt werden.
- Glutathionperoxidase (GPx)
Anhand von GPx kann man feststellen, auf welchem Niveau der Abbau von lipid- und lipoïdbedrohenden ROS zum Schutz der Zellmembranen stattfindet. GPx ist u. a. eine Gegenspielerin der reaktiven Sauerstoffverbindung Wasserstoffperoxid.
In einer im Jahre 2005 durchgeführten Post-Mortem-Studie unter Führung der Universität Würzburg untersuchten Tanja M. Michel und ihr Team den präfrontalen Cortex und die Hippocampi 14 Verstorbener. Bei der Hälfte der Personen wurde zu Lebzeiten eine Depression diagnostiziert, die andere Hälfte war zu Lebzeiten neuropsychiatrisch unauffällig. Die in die Untersuchung einbezogenen Personen starben überwiegend im höheren und hohen Alter zwischen dem 61. und 93. Lebensjahr, nur eine Person starb vor Vollendung des 31. Lebensjahres.
Es wurde untersucht, ob in affektrelevanten Hirnarealen Depressiver erhöhter oxidativer Stress gemessen werden kann (Quelle: T. M. Michel, S. Frangou, R. Zoechling et al., Evidence for oxidative stress in the frontal cortex in patients with recurrent depressive disorder - a postmortem study, Psychiatry Research 151/2007, Elsevier Ireland Ltd., http://www.psy-journal.com/article/...). Als Parameter wurden die Konzentrationen zweier Superoxid-Dismutasen ‑ Kupfer-Zink-SOD und Mangan-SOD ‑ in den jeweiligen Hirnarealen gemessen, die beide der Neutralisierung und Entfernung von Sauerstoffradikalen dienen. Kupfer-Zink-SOD spielen vor allem in Gliazellen eine Rolle, zum Beispiel in Oligodendrozyten, Mangan-SOD kommen hier nur in geringen Mengen vor. Umgekehrt verhält es sich bei der Mangan-SOD ‑ sie findet man hauptsächlich in Nervenzellen und weniger in Gliazellen.
Michel und ihr Team verweisen in ihrem Studienreport zunächst auf die Fülle von Studienergebnissen, die Zusammenhänge zwischen der Erkrankung an einer rezidivierenden depressiven Störung, Volumenminderungen des präfrontalen Cortex und der Hippocampi sowie Neuronen- und Gliazellenveränderungen als wahrscheinlich erscheinen lassen: „Structural brain imaging studies of patients with DD [Anmerkung: DD = Depressive Disorder] have consistently reported volumetric reductions in the prefrontal cortex (PFC) and the hippocampus (Campbell et al., 2004). Postmortem studies have revealed striking reductions in glial cell number and density in these brain regions as well as more subtle changes in neuronal density and size (Rajkowska et al., 1999, 2001; Cotter et al., 2001, 2002; Bowley et al., 2002; Harrison, 2002).“ (Quelle: wie oben). Die Studienautoren erwähnen auch, dass noch kein Mechanismus identifiziert wurde, der eine solche Entwicklung verursacht haben könnte („At present, no specific pathophysiological process has been linked with these morphological changes.“).
Die Thematik von Neuronen- und Volumenveränderungen affektrelevanter Hirnareale wurde im ersten Kapitel erörtert und mit Studien belegt. Die bei Tanja M. Michel angeführten Beobachtungen derartiger Phänomene stützen einmal mehr die hier zur Diskussion stehenden zentralen kausaltheoretischen Thesen (→ Abschnitte 1.3 bis 1.6).
Obwohl die Arbeit Untersuchungen an Hirnpräparaten von lediglich 14 Personen umfasste und die Ergebnisse kein endgültiges Urteil erlauben, sind deren Resultate dennoch bemerkenswert. So war die Konzentration der Kupfer-Zink-SOD im präfrontalen Cortex depressiver Personen signifikant erhöht. In den Hippocampi waren sie ebenfalls erhöht, jedoch nicht signifkant zur Vergleichsgruppe. Die Mangan-SOD war nirgends erhöht. Die Autoren der Studie zogen den Schluss, dass oxidativer Stress vor allem in den Gliazellen des präfrontalen Cortex eine Rolle spielt und die erhöhte Menge an Kupfer-Zink-SOD eine Reaktion auf diesen Stress darstellt. Davon betroffen, so die Autoren, wären vor allem die als besonders anfällig für oxidativen Stress geltenden Oligodendrozyten. Aber auch Astrozyten, eine andere Sorte von Gliazellen, deren Empfindlichkeit für oxidativen Stress mit dem Alter steigt, stehen durch die Studie im Verdacht, eine erhöhte Menge Kupfer-Zink-SOD zu enthalten. Astrozyten spielen bei der Regeneration von Hirnzellen und Neurogenese eine wichtige Rolle.
Dass es in den Hippocampi nicht zu einer Messung erhöhter SOD-Werte gekommen ist, bedeutet übrigens nicht, dass hier oxidativer Stress keine Rolle spielt. Im Gegenteil könnte eine geringere Konzentration zelleigener Superoxid-Dismutasen darauf hindeuten, dass der Abwehrmechanismus nicht mehr adäquat reagiert und gerade hier die oxidative Schädigung hoch gewesen sein könnte. Eine solche Vermutung ist jedoch nur durch eine Vermessung der Hippocampi weiter verifizierbar, die in dieser Studie aber (leider) nicht erfolgte.
Denn die eigentliche Intention des Teams um Tanja Michel war die Beantwortung der Frage, ob oxidativer Stress zu Struktur- bzw. Zellveränderungen im Gehirn geführt haben könnte oder daran zumindest mitbeteiligt ist. Im Abstrakt des Studienreports heißt es dazu: „Here we examine the possibility that oxidative stress, widely implicated in neuronal cell damage, may contribute to these brain structural changes.“ (Quelle: wie oben). Über eine Vermessung der Volumina der verwendeten Gehirne oder sonstige strukturelle Untersuchungen der Gewebeproben wurde allerdings nichts berichtet.
Ihre Studienergebnisse veranlassten die Autoren auch zu der Einschätzung, dass sie zur wachsenden Zahl von Hinweisen beigetragen haben, welche oxidativen Stress als auslösenden Faktor in der Pathophysiologie der Depression identifizieren: „Our findings contribute to the growing body of evidence implicating oxidative stress in the pathophysiology of depressive disorder.“ (Quelle: wie oben).
Mit einer aufwändigen Literaturrecherche werteten die australischen Forscher Ng, Berk, Dean und Bush an der Universität Melbourne bis Ende 2007 eine Vielzahl von Studien aus, die verschiedene potentielle Zusammenhänge zwischen psychiatrischen Erkrankungen und oxidativem Stress analysierten. Dabei wurde der Fokus auf die Ursachen von Schizophrenie, der Bipolaren Störung und der Major Depression gelegt, in geringerem Umfange auch auf Angst- und andere psychiatrische Erkrankungen, wobei hier ausschließlich die Erkenntnisse über die beiden Affekterkrankungen diskutiert werden. In der Referenzenliste der Literaturstudie sind 43 Veröffentlichungen aufgeführt (Quelle: Felicity Ng, Michael Berk, Olivia Dean, Ashely I. Bush, Oxidative stress in psychiatric disorders: evidence base and therapeutic implications, International Journal of Neuropsychopharmacology, 11/2008, https://ijnp.oxfordjournals.org/...).
In den Studien zur Bipolaren Störung und Major Depression wurden verschiedene Substanzen im Blutserum oder -plasma analysiert. Darunter befanden sich das Oxidant Stickstoffmonoxid (NO), zelleigene Antioxidantien (Superoxid-Dismutasen, Catalase, Glutathionperoxidase), externe Antioxidantien (Vitamin C und E u. a.) oder Biomarker für oxidativen Stress (TBARS, Malondialdehyd, 4‑Hydroxynonenal, 8‑Hydroxydesoxyguanosin). Auch Genstudien, pharmakologische Studien mit Untersuchungen antioxidativer Eigenschaften etablierter Pharmazeutika, präklinische Studien mit Tieren bzw. In-vitro-Zellstudien und eine klinische Studie über die potentielle Wirkung des Radikalfängers Acetylcystein (NAC), einer Vorstufe von Glutathion, wurden in den Vergleich einbezogen.
Das Team um Felicity Ng stellte in seiner Zusammenfassung der Studienrechere fest, dass alle Resultate eine solide Grundlage für die Hypothese bilden, oxidativer Stress spiele eine Rolle bei der Genese der Bipolaren Störung und Depression (und anderer psychiatrischer Erkrankungen) als ein universeller pathogener Mechanismus mit der Chance, auf dieser Basis neuartige therapeutische Maßnahmen zu entwickeln: „For bipolar disorder and depression, a solid foundation for oxidative stress hypotheses has been provided by biochemical, genetic, pharmacological, preclinical therapeutic studies and one clinical trial. (…) In conclusion, multi-dimensional data support the role of oxidative stress in diverse psychiatric disorders. These data not only suggest that oxidative mechanisms may form unifying common pathogenic pathways in psychiatric disorders, but also introduce new targets for the development of therapeutic interventions.“ (Quelle: wie oben).
Im ausführlicheren Diskussionsabschnitt des Abschlussdokuments werden in Bezug auf die Bipolare Störung und die Major Depression die wichtigen Studienerkennntnisse aufgezählt:
- Es gibt Beweise einer erhöhten Lipidperoxidation bei beiden Affektstörungen.
- Bestimmte körpereigene Antioxidatien, beispielsweise Albumin und Bilirubin, sind bei einer Depression reduziert.
- Die Ergebnisse von Einzelstudien über die Rolle des Stickstoffmonoxids (NO) und körpereigenen antioxidativen Enzymen sind zwar nicht konsistent, aber es gibt Hinweise auf deren Beteiligung an der Entstehung affektiver Erkrankungen.
- Die Ergebnisse von Gen- und Pharmastudien weisen darauf hin, dass oxidativer Stress bei der Pathogenese u. a. der Bipolaren Störung eine Rolle spielt.
- Bestimmte Stimmungsaufheller und Antidepressiva scheinen eine antioxidative Wirkung zu entfalten.
- Die Anwendung des Radikalfängers Acetylcystein (NAC) scheint bei der Bipolaren Störung einen Nutzen zu haben.
Keine auf einer objektiv belegbaren Grundlage beruhenden Aussage findet sich in den analysierten Studien zur Frage, auf welchen Ursachen oxidativer Stress beruhen könnte. Nach Meinung der Autoren komme eine übermäßige mitochrondriale Energieerzeugung, dysfunktionale Schutzmechanismen oder beides infrage.
Einige Details einer Studie über die Anwendung des Radikalfängers Acetylcystein (NAC), einer Vorstufe des Antioxidants Glutathion, sind hier noch erwähnenswert. Durchgeführt wurde sie von einem Team um Michael Berk von The Mental Health Research Institute of Victoria, der auch bei der Übersichtsstudie unter der Leitung von Felicity Ng beteiligt war (Quelle: M. Berk et al., N-acetyl cysteine for depressive symptoms in bipolar disorder ‑ a double-blind randomized placebo-controlled trial, Biological Psychiatry, 9/2008, Society of Biological Psychiatrie, Elsevier, New York, USA 2008, http://www.ncbi.nlm.nih.gov/....). Untersucht wurde die Wirkung einer NAC-Gabe (zweimal täglich ein Gramm) bei unveränderter Einnahme von Psychopharmaka auf die depressive Symptomatik der Bipolaren Störung. Während der Einnahme besserte sich diese deutlich, während sich nach der vollständigen Ausschleichung der Substanz über drei Wochen der ursprüngliche schlechte Zustand wieder einstellte. Man kann daraus schließen, dass NAC ein wirksames Mittel zur Verbesserung der Symptomatik bei der Bipolaren Störung ist. Ebenfalls legt das Ergebnis den Schluss nahe, dass oxidativer Stress bei der Pathogenese der Erkrankung eine kausale Rolle spielt.
Im Februar 2007 erschienen in der Online-Bibliothek von Wiley-Blackwell die Ergebnisse einer Studie, die unter der Leitung von Aslı Sarandöl und einem Team der Fakultät für Medizin der Universität in Uludag im türkischen Bursa durchgeführt wurde (Quelle: A. Sarandöl et al., Major depressive disorder is accompanied with oxidative stress: short-term antidepressant treatment does not alter oxidative-antioxidative systems, Human Psychopharmacology: Clinical and Experimental, Wiley-Blackwell, Oxford/U. K. 2007, http://onlinelibrary.wiley.com/...). Die Ziele der Studie waren Untersuchungen zum oxidativen Status der Beteiligten und eine Antwort auf die Frage, ob Antidepressiva hierauf einen Einfluss haben.
Sarandöl und ihr Team untersuchten mehrere Substanzen und Marker oxidativen Stresses bei 96 Patienten mit einer Major Depression und einer Vergleichsgruppe aus 54 gesunden Probanden. Es ging dabei um die Blutplasma- bzw. Blutserumwerte von Malondialdehyd (MDA), Vitamin E, Vitamin C, Carotinioiden oder Glutathionperoxidase (GPx). Ebenfalls wurde die Konzentration von Superoxid-Dismutasen (SOD) der Erythrozyten gemessen. Die Werte wurden vor und nach einer sechswöchigen Therapie mit Antidepressiva bestimmt.
Die Resultate stützen die Hypothese eines Zusammenhangs zwischen oxidativem Stress und einer Depression. Die Malondialdehyd-Werte des Blutplasmas erkrankter Studienteilnehmer waren gegenüber den gesunden Probanden signifkant erhöht, ebenfalls die Anfälligkeit der Erythrozyten für oxidativen Stress. Die Aktivitäten der SOD waren bei den erkrankten Personen nicht nur deutlich erhöht, ihre Intensität korrelierte auch positiv zur Schwere der Depression.
Die sechswöchige Einnahme von Antidepressiva hatte keinerlei Auswirkungen ‑ weder auf die oxidativen Prozesse noch auf das antioxidative System.
Auch die Ergebnisse einer Meta-Analyse aus dem Jahre 2013 von Priya Palta und ihrem Team der University of North Carolina at Chapel Hill in den USA, die 23 zwischen Januar 1980 und Dezember 2012 durchgeführte Studien mit insgesamt 4.980 Studienteilnehmern über etwaige Zusammenhänge zwischen einer Depression und oxidativem Stress bzw. einer Depression und dem antioxiativen Status der Teilnehmer einschloss, deuten auf einen Zusammenhang beider Merkmalskombinationen hin (Quelle: Priya Palta, Laura J. Samuel et al., Depression and Oxidative Stress: Results From a Meta-Analysis of Observational Studies, Psychosomatic Medicine, 1/2014, American Psychosomatic Society, Lippincott Williams & Wilkins/Wolters Kluwer, Philadelphia, USA 2014, http://www.ncbi.nlm.nih.gov/...).
Als statistische Messgröße wurde die Effektstärke nach Cohen (Cohens d) berechnet, wobei sich ein relativ hoher Wert von d = 0,55 ergab für einen positiven Zusammenhang zwischen den beiden Merkmalen Depression und oxidativem Stress bzw. ein negativer Zusammenhang von d = ‑0,24 für Depression und antioxidativem Status.
Aus allen in die Meta-Analyse eingeschlossenen Studien ergab sich maximal ein korrelativer Zusammenhang zwischen beiden Merkmalskombinationen. Eine Interpretation der Ergebnisse über kausale Beziehungen wäre daher spekualtiv, beruhte nach den Ergebnissen jedoch zumindest auf harten Tatsachen.
Interessanterweise wird in der Zusammenfassung der Studieninhalte der Palta-Studie gleich zu Beginn erwähnt, dass die Meta-Analyse die Hypothese erhärten oder widerlegen soll, eine Depression führe zu vermehrtem oxidativen Stress bzw. zu einem erniedrigten antioxidativen Status, was ja ein im Vergleich mit der Kausaltherorie umgekehrter Kausalzusammenhang wäre.
Diese spezielle Sicht ist zwar nicht zu beanstanden und steht auch nicht mit den kausaltheoretischen Annahmen im Widerspruch, lenkt aber aus kausaltheoretischer Sicht von dem naheliegenden Szenario eher ab. Die Studienergebnisse eignen sich genauso gut als begründete spekulative Grundlage dafür, dass erhöhter oxidativer Stress eine Depression triggert, so wie dies auch in der Michel-Studie in Übereinstimmung mit der kausaltheoretischen Sichtweise gemacht wurde (dort über die Kausalkette oxidativer Stress → strukturelle Hirnveränderungen → Affektstörung).
Andererseits liefert die zu überprüfende Hypothese der Palta-Studie wiederum aus kausaltheoretischer Sicht interessante Aspekte: Erhöht eine Affekterkrankung tatsächlich ihrerseits oxidativen Stress im Gehirn, könnte das auf einen selbstverstärkenden Effekt der Erkrankung hinweisen, denn eine erhöhte Oxidation würde das Gehirn zusätzlich schädigen.
Ein andere Meta-Analyse aus dem Jahre 2014 des EMGO Institute for Health and Care Research der Universität Amsterdam und eines Teams um die Niederländische Psychiaterin Brenda Pennix (Quelle: C. Black, B. Pennix et al., Is depression associated with increased oxidative stress? A systematic review and meta-analysis, Psychoneuroendocrinology, 1/2015 (Vol. 51), Elsevier B.V., Niederlande, Amsterdam 2015, http://www.ncbi.nlm.nih.gov/...) umfasste 18 Einzelstudien. Ziel war es, Zusammenhänge zwischen dem Auftreten von Depressionssymptomen (DS), einer Major-Depression (MDD) oder einer Bipolaren Störung (BD) mit 8‑Hydroxydesoxyguanosin, dem Marker oxidativer DNA-Schäden und F2‑Isoprostan, einem Marker für die Stärke der Lipidperoxidation, aufzuspüren. Die Merkmale DS, MDD und BD wurden gebündelt analysiert.
Mit Hilfe der Hedges-Effektstärke g wurde ein Zusammenhang zwischen DS, MDD und BD einerseits und oxidativem Stress andererseits bestätigt. Der Wert für 8‑Hydroxydesoxyguanosin betrug g = 0,31, für F2‑Isoprostan g = 0,48.
In der zusammenfassenden Schlussfolgerung am Ende der Studiendokumentation wurden keine weiteren Vermutungen über Kausalitäten geäußert. Aus kausaltheoretischer Sicht ist diese Meta-Studie ein weiterer starker Hinweis dafür, dass oxidativer Stress als (Mit-)Verursacher einer Affektstörung infrage kommen könnte.
Fazit: Oxidativer Stress der endogenen Zellatmung als Verursacher Affektiver Störungen
Oxidativer Stress resultiert aus einer übermäßigen Anzahl hoch aggressiver reaktiver Sauerstoff-Spezies (ROS), die eine Zelle mit ihren antioxidativen Schutzmechanismen nicht mehr neutralisieren und beseitigen kann. Besonders während der wichtigen Energieversorgungsprozesse in der Atmungskette entsteht potentiell oxidativer Stress.
Oxidativer Stress kann sämtliche Zellstrukturen - Lipoide und Proteine einschließlich der DNA - schädigen mit der Folge unterschiedlicher Probleme: von leichten über schwere Prozessstörungen bis hin zum Zelltod oder zu Tumor- bzw. Krebszellen.
Aus nicht reparierten DNA-Schäden werden somatische Mutationen und auf nachfolgende Zellgenerationen übertragen, was die Proteinbiosynthese langfristig behindern kann. Kommen im Laufe der Zeit weitere Mutationen hinzu (Akkumulation), reduziert sich die Qualität der Proteinbiosyntheseprozesse weiter. Das hat Auswirkungen auf sämtliche Zell- und Organfunktionen, einschließlich der antioxidativen Schutzprozesse, deren Qualität immer weiter abnimmt. Oxidativer Stress hat daher mit hoher Wahrscheinlichkeit einen großen Anteil am Alterungsprozess von Zellen und Organen.
Besonders das Gehirn ist von oxidativem Stress bedroht. Zum einen liegt das an seinem hohen Energiebedarf mit entsprechend hohem ROS-Aufkommen. Auch der hohe Fettgehalt macht das Gehirn anfällig für die Lipidperoxidation besonders der inneren und äußeren Zellmembranen. Nervenzellen und bestimmte Gliazellen, zum Beispiel Oligodendrozyten, haben darüber hinaus ein nur begrenzt arbeitendes antioxidatives Schutzsystem und der Tod von Hirnzellen ist unter normalen Umständen in vielen Hirnarealen nicht rückgängig zu machen.
Für die Pathogenese Affektiver Störungen ist oxidativer Stress in affektrelevanten Hirnarealen demnach besonders relevant. Je höher oxidativer Stress ist, desto größer ist das Risiko der Entstehung oder Verstärkung einer Erkrankung.
Studien zu oxidativem Stress und Affekterkrankungen stellten erhöhte Werte bei Erkrankten fest. Daraus ergibt sich zwar kein Nachweis eines Kausalzusammenhangs zwischen oxidativem Stress und Affektiven Störungen, die Annahme eines solchen ist aber plausibel.
Aus kausaltheoretischer Sicht besteht im Zentralnervensystem bzw. Gehirn folgender einfacher Zusammenhang zwischen oxidativem Stress und Affektiven Störungen:
Oxidativer Stress → Zellprozessänderungen → Hirnstrukturveränderungen → Affektstörung
Eine solche Sichtweise hat sich allerdings bis zum heutigen Tage in der Psychiatrie nicht durchgesetzt, obwohl aufgrund vieler darauf hindeutender Studienergebnisse kausale Zusammenhänge zwischen oxidativem Stress und Affekterkrankungen zumindest von einigen Wissenschaftlern und Medizinern mittlerweile als wahrscheinlich erachtet werden.
Ein Team um die Würzburger Psychiaterin Tanja Michel stellte im Jahre 2005 in einer Post-Mortem-Studie erhöhten oxidativen Stress in affektrelevanten Hirnarealen Verstorbener mit einer zu Lebezeiten diagnostizierten Depression fest und geht von aus, dass es sich hier um einen Kausalzusammenhang handelt.
Eine Studie über die Anwendung eines Antioxidans ergab, dass sich damit die depressive Symptomatik einer Bipolaren Störung verbesserte, was ebenfalls für einen kausalen Zusammenhang zwischen oxidativem Stress und einer Affektstörung spricht.
Es ist interessant, dass auch ein umgekehrter Kausalzusammenhang in Erwägung gezogen wird. So argumentieren Palta und ihr Team aufgrund von Studienergebnissen, eine Depression selber führe möglicherweise zu einem höheren oxidativen Stress. Ein Nachweis dürfte jedoch schwierig werden. Diese Vermutung steht auch nicht im Widerspruch zu kausaltheoretischen Annahmen, denn ein solcher Zusammnhang wird nicht explizit ausgeschlossen und könnte bedeuten, dass eine Affekterkrankung einen selbstverstärkenden Faktor hat.
4.13.7 Gift- und Schadstoffe (ohne Suchtstoffe) ▲
Im täglichen Leben kommt jeder unausweichlich mit Gift- und Gefahrstoffen in Kontakt. Manche reizen vorwiegend Haut, Atemwege oder Schleimhäute, andere führen zu zahlreichen Beschwerden, von denen grundsätzlich jeder Bereich des Körpers betroffen sein kann, insbesondere auch das Nervensystem. So greifen beispielsweise gerade Insektizide oft die Nerven der Tiere mit dem Ziel an, sie zu töten - und das macht diese Substanzen auch für Menschen zu einer erheblichen Gefahr.
Schadstoffbedingten Störungen des menschlichen Nervensystems folgen häufig neurologische und psychische Symptome. Im Jahre 2006 wurden in einer Veröffentlichung der Universität Süd-Dänemark in Odense etwa 200 Substanzen als neurotoxisch für Menschen identifiziert (Quelle: P. Grandjean, P. J. Landrigan, Developmental neurotoxicity of industrial chemicals, Institute of Public Health, University of Southern Denmark/Odense, The Lancet, 368 (9553), 12/2006, Elsevier/U. K., London 2006, www.http://www.sciencedirect.com/...). Dies kann jedoch nur die Spitze des Eisbergs sein, denn die Anzahl aller Industriechemikalien liegt im hohen fünfstelligen Bereich.
Es muss nicht unbedingt eine große Substanzenmenge einwirken, damit es zu einer Vergiftung kommt - viele Stoffe sind auch in sehr niedrigen Dosen giftig und können sich darüber hinaus im Körper anreichern. Bei einigen Substanzen, beispielsweise Verbindungen aus der Gruppe polychlorierter Biphenyle (PCB), ist die akute Toxizität mit höheren Mengen sogar eher gering, während deren chronische Toxizität vergleichsweise schon bei minimalen Mengen sehr hoch ist.
Gesundheitsrisiken einzelner Schadstoffe können häufig nicht vollständig abschließend bewertet werden, da neben bekannten auch unbekannte Schadwirkungen nicht auszuschließen sind. Selbst bei intensiv verwendeten, weit verbreiteten Substanzen, wie zum Beispiel Glyphosat, herrscht immer noch völlige Uneinigkeit in der Frage, ob und wie gefährlich sie sind.
Industriegifte
Die meisten Industriegifte werden bei der Herstellung und Verwendung von Kunststoffen, Farben und Lacken, Kraftstoffen (Benzin, Diesel), Holzschutz-, Pflanzenschutz- und Reinigungsmitteln produziert sowie bei der Schädlings- bzw. Insektenbekämpfung einschließlich der Pilz- und Sporenbekämpfung (Fungizide) und der Unkrautvernichtung.
Durch die im Jahre 2000 initiierte Stockholmer POP-Konvention, eine multinationale Übereinkunft über das Verbot persistenter (= dauerhafter) organischer Schadstoffe, ist die Herstellung und Verwendung von derzeit etwa 30 Substanzen verboten oder stark eingeschränkt (Quelle: Umweltbundesamt zur Stockholm-Konvention, http://www.umweltbundesamt.de/...).
In Tabelle 23 sind häufig eingesetzte Industriegifte und eine Auswahl ihrer neurotoxischen Wirkungen auf das zentrale und periphere Nervensystem (ZNS, PNS) aufgelistet. Aufgrund der hohen Anzahl vieler verschiedener Substanzen und Wirkungen kann sie nur beispielhaft sein.
TABELLE 23: EINE AUSWAHL NEUROTOXISCHER INDUSTRIEGIFTE
Schadstoff(-gruppe) |
Verwendungsbeispiele, Regelement |
Beispiele neurologischer Symptome |
|---|---|---|
| Blei(-verbindungen) | Bauindustrie, insbesondere Rohre Benzinzusatzstoff, reglementiert Elektrotechnik Farben Maschinenbau Pflanzenschutzmittel, reglementiert |
ZNS-Schädigungen (u. a. Enzephalopathie) |
| Bioallethrin | Insektizid (Anti-Mücken-Sprays) Pflanzenschutzmittel |
Krämpfe PNS-Schädigungen ZNS-Schädigungen |
| Chlorpyrifos | Insektizid | Bewusstseinsstörungen Kopfschmerzen Krämpfe PNS-Schädigungen Sehstörungen Sprachstörungen Tremor Verwirrtheit Zentrale Atemlähmung ZNS-Schädigungen |
| Cypermethrin | Insektizid Holzschutzmittel |
Kopfschmerzen PNS-Schädigungen Schwindel |
| DDT | Insektizid, reglementiert Holzschutzmittel, verboten POP-Konvention |
Erregbarkeit (bei niedrigen Dosen) Lähmungen (bei höheren Dosen) ZNS-Schädigungen |
| Diazinon | Insektizit | Bewusstseinsstörungen Krämpfe PNS-Schädigungen Sprachstörungen Tremor Verwirrtheit Zentrale Atemlähmung |
| Endosulfan | Insektizid, reglementiert Holzschutzmittel, reglementiert POP-Konvention |
Bewusstseinsstörungen Kopfschmerzen Koordinationsstörungen Krämpfe Schwindel ZNS-Schädigungen |
| Etyhlacetat | Aromastoffgewinnung Extraktionsmittel (z. B. Entkoffeinierung) Klebstoffe Kosmetika Lacke Parfumherstellung Missbrauch als sog. „Schnüffelstoff“ |
Narkotisierend bei höheren Dosen ZNS-Schädigungen |
| Fenitrothion | Insektizid, Verbot in EU + Schweiz | Bewusstseinssstörungen Krämpfe Parästhesien PNS-Schädigungen Sprachstörungen Tremor Verwirrtheit Zentrale Atemlähmung ZNS-Schädigungen |
| Formaldehyd | Bauwirtschaft Farben Lacke Pharmazie (Desinfektion) Holzverarbeitung Kosmetika Kunststoffherstellung Möbelindustrie Reinigungsmittel Tabak Textilverarbeitung |
Gedächtnisstörungen Konzentrationsstörungen Schlafstörungen ZNS-Schädigungen |
| Glyphosat | Unkrautvernichtung | Autismus (Verdacht) ZNS-Schädigungen (indirekt, Verdacht) |
| Hexachlorcyclohexan [synonym: Technisches HCH*] |
Insektizit, seit 1977 Verbot in West-DE POP-Konvention für α-HCH |
Kopfschmerzen Schwindel ZNS-Schädigungen |
| γ-Hexachlorcyclohexan [synonym: Lindan] |
Insektizid, stark reglementiert Anti-Läuse-Shampoos Holzschutzmittel, verboten POP-Konvention |
Cerebrale Blutflussstörungen Ermüdbarkeit Gedächtnisstörungen Gleichgewichtsstörungen Konzentrationsstörungen Kopfschmerzen Lähmungen Multiple Sklerose Parkinsonismus Reizbarkeit Schwindel Tremor Verwirrung ZNS-Schädigungen |
| n-Hexan | Farben Klebstoffe Kunststoffherstellung Lacke |
Apathie Bewusstseinsstörungen Kopfschmerzen PNS-Schädigungen Schwindel ZNS-Schädigungen |
| Methylbromid [synonym: Brommethan] |
Begasungsmittel Pflanzenschutzmittel |
Halluzinationen Koordinationsstörungen Krämpfe Lähmungen Verwirrung ZNS-Schädigungen |
| (Oxy-)Chlordan | Holzschutzmittel Insektizid Pflanzenschutzmittel POP-Konvention |
Krämpfe Tremor Verwirrung |
| Pentachlorphenol (PCP) | Holzschutzmittel (gegen Pilzbefall) Lederimprägniermittel Textilimprägniermittel POP-Konvention |
Antriebslosigkeit Bewusstseinsstörungen Cerebrale Blutflussstörungen Erschöpfung Gedächtnisstörungen Kopfschmerzen Libidostörungen Müdigkeit Schlafstörungen Schwindel ZNS-Schädigungen |
| Pentan | CKW-Ersatzmittel Kältemittel in Kühlschränken Klimaanlagen |
Bewusstseinsstörungen Kopfschmerzen Schwindel ZNS-Schädigungen |
| Perchlorethylen (PER) | Chemische Textilreinigung Textilimprägniermittel |
Schädigungen des dopaminergen Systems Gedächtnisstörungen Konzentrationsstörungen Koordinierungsstörungen Kopfschmerzen Kurzzeitgedächtnisstörungen Müdigkeit Narkotisierend Reizbarkeit Sprachstörungen ZNS-Schädigungen |
| Permethrin | Insektizid Holzschutzmittel Pflanzenschutzmittel Pharmazie Textilindustrie |
Bewusstseinsstörungen Gedächstnisstörungen Intelligenzminderung Konzentrationsstörungen Kopfschmerzen Krämpfe Parästhesien Schwindel Sprachstörungen ZNS-Schädigungen |
| Polychlorierte Biphenyle (PCB) |
Energieversorgung Haushaltsgeräte Kunststoffherstellung Lacke POP-Konvention |
Aggressives Verhalten Aufmerksamkeitsstörungen Schädigungen des dopaminergen Systems Gedächtnisstörungen Hyperaktivität Intelligenzminderung Kopfschmerzen Lernstörungen Schlafstörungen Schwindel Störungen der geistigen Entwicklung ZNS-Schädigungen |
| Phosphorsäureester | Insektizid Kunststoffherstellung Lacke Pflanzenschutzmittel Reinigungsmittel |
Kopfschmerzen Krämpfe Parästhesien PNS-Schädigungen Sehstörungen Sprachstörungen |
| Pyrethroide | Insektizid Pflanzenschutzmittel Pharmazie |
Bewusstseinsstörungen Gedächtnisstörungen Konzentrationsstörungen Krämpfe Parästhesien ZNS-Schädigungen |
| Quecksilber | Batterieherstellung Farben Kunststoffherstellung Rohstoffindustrie Zahnmedizin (Amalgam) |
PNS-Schädigungen ZNS-Schädigungen |
| 1,1,1-Trichlorethan | Farben Klebstoffe Reinigungsmittel Missbrauch als sog. „Schnüffelstoff“ |
Bewusstseinsstörungen Koordinierungsstörungen Kopfschmerzen Schwindel ZNS-Schädigungen |
| Trichlorethen (TRI) [synonym: Trichlorethylen] |
Bitumenherstellung Chemische Textilreinigung Extraktionsmittel (z. B. Entkoffeinierung) Farben Reinigungsmittel Missbrauch als sog. „Schnüffelstoff“ |
Bewusstseinsstörungen Gedächstnisstörungen Schädigungen des dopaminergen Systems Konzentrationsstörungen Kopfschmerzen Schwindel ZNS-Schädigungen |
Tabelle 23: Die Herstellung oder Verwendung der Substanzen mit dem Hinweis auf die POP-Konvention (für die englische Bezeichnung „persistent organic pollutants“) ist im Rahmen des Stockholmer Übereinkommens über persistente (= dauerhafte) organische Schadstoffe entweder weltweit verboten oder stark eingeschränkt (Stand Mai 2013). Darüber hinaus gibt es für einige Substanzen nationale Regelungen. Alle Angaben zur Verwendung und über neurologische Symptome sind beispielhaft.
* Technisches HCH besteht aus fünf HCH-Verbindungen, wobei das α-HCH mit ca. 70% den größten Anteil hat (Quelle: Stoffbericht Hexachlorcyclohexan/HCH, in: Handbuch Altlasten und Grundwasserschadensfälle, Zentraler Fachdienst Wasser-Boden-Abfall-Altlasten bei der Landesanstalt für Umweltschutz Baden-Württemberg, Karlsruhe 1993).
Weitere Schadstofftypen: NO2, Staubpartikel und Mikroplastik
Darüber hinaus sind hier noch Schadstoffe aus Verbrennungsprozessen relevant, wobei Stickstoffdioxid (NO2), Feinstaub (<10 µm Ø), lungengängiger Feinststaub (< 2,5 µm Ø) bzw. ultrafeine Partikel (< 0,1 µm Ø) unterschieden werden. Ultrafeine Partikel sind in der Lage, Zellmembranen zu passieren und gelangen so u. a. in den Blutkreislauf, während lungengängiger Feinststaub maximal in die Lungenbläschen (Alveolen) eindringen kann.
Auch potentielle Folgen der verstärkten Mikroplastikbelastung auf das Zentralnervensystem wird thematisiert. Erste Studien weisen darauf hin, dass Mikroplastik die Blut‑Hirn‑Schranke übernwinden kann und damit eine potentielle Ursache neurodegenerativer Erkrankungen sein kann.
Mechanismen der Neurotoxizität von Gift- und Schadstoffen
Die neurologischen Mechanismen neurotoxisch wirkender Substanzen werden nur teilweise, manchmal noch gar nicht verstanden. Von einigen Stoffen kennt man deren morphologisch-strukturelle oder funktionale Ziele im Nervensystem. Einige Toxine wirken mutagen und können DNA-Veränderungen herbeiführen (z. B. Asbest oder PCB).
Details hierzu sind nicht von Bedeutung. Es genügt eine kurze systematische Übersicht, um sich des hohen Risikos dieser Substanzen und ihrer vielen Möglichkeiten bewusst zu werden, Nervenzellen nachhaltig zu schädigen. Die neurotoxische Wirkung einer Substanz kann direkt oder indirekt erfolgen und sowohl Strukturen als auch Funktionen der Nerven- und Gliazellen angreifen. Folgende Angriffsziele werden unterschieden:
- Nervenzellkörper (Neuronopathie)
- Axon (Axonopathie)
- Myelinscheiden (Myelinopathie)
- Synapsen
Die Schadenintensität hängt hauptsächlich von zwei Faktoren ab:
- Einwirkungszeit (Verweildauer)
Je länger die Wirkungsdauer, desto stärker die Wirkung ‑ von dieser Regel gibt es keine Ausnahme.
- Gewebekonzentration
Man kann davon ausgehen, dass eine größere Menge auch einen größeren Schaden anrichtet: Die Dosis macht das Gift. Es wurde aber schon darauf hingewiesen, dass geringe Dosen bestimmter Substanzen in Verbindung mit einer längeren Einwirkzeit höhere langfristige Schäden zur Folge haben können als kurzzeitige hohe Giftmengen, beispielsweise bei der Gruppe der polychlorierten Biphenyle (PCB).
Weitere Einflussfaktoren auf die Neurotoxizität von Gift- und Schadstoffen
Ebenfalls spielen Einflüsse eine Rolle, die Aufnahmepfade oder Entgiftungs-, Reparatur- und Schutzprozesse der Nerven- und Gliazellen bzw. des restlichen Körpers erheblich beinflussen:
- Alter
- (Poly-)Genetische Prädispositionen
- Erkrankungen des Körpers
- Psychiatrisch-neurologische Erkrankungen
- Zustand der Blut-Hirn-Schranke
- Ernährungszustand mit Makro- und Mikronährstoffen
- Zustand der Leber (Qualität der Entgiftung über die Leber)
- Zustand der Nieren (Qualität der Entgiftung über die Nieren)
- Schlafqualität (→ folgender Abschnitt über das Glymphatische System)
Auch die Funktionsfähigkeit der Schilddrüse nimmt Einfluss: Eine gift- oder schadstoffbelastete Schilddrüse kann zu einer Organunterfunktion führen mit entsprechenden negativen Auswirkungen auf die Gemütslage.
Die Entgiftung des Gehirns durch das Glymphatische System
Im Jahre 2012 entdeckten Forscher der Medizinischen Fakultät der Universität in Rochester/USA einen Entsorgungsmechanismus für Schadstoffe im Gehirn. Ein Jahr später veröffentlichten Forscher derselben Universität eine weitere Studie, deren Ergebnisse darauf hindeuten, dass dieser Entsorgungsvorgang insbesondere während der Schlafphase erfolgt (Quellen: J. J. Iliff, M. Nedergaard et al., A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β, Science Translational Medicine, 4 (147), 8/2012, Washington, D. C., USA 2012, http://stm.sciencemag.org/... und L. Xie, M. Nedergaard et al., Sleep Drives Metabolite Clearance from the Adult Brain, Science, 342 (6156), 10/2013, Washington, D. C., USA 2013, http://science.sciencemag.org/...).
Aufgrund der wesentlichen Beteiligung der Gliazellen an den Entsorgungsprozessen sprechen die Entdecker von einem Glymphatischen System in Anlehnung an den Begriff des Lymphsystems. Es handelt sich um ein Netz kleiner Kanäle zwischen Nerven- und Gliazellen, mit deren Hilfe die Giftstoffe während des Schlafs über die Hirnflüssigkeit ausgeschwemmt werden.
Zwar wurden zunächst Prozesse identifiziert, bei denen zelleigene Stoffwechselprodukte entsorgt werden. Es ist jedoch möglich und auch wahrscheinlich, dass über diesen Mechanismus ebenfalls Toxine, die von außen in das Gehirn gelangt sind, wieder ausgeschieden werden. Das gilt auch für deren giftige Abbauprodukte. Sie würden durch ein intaktes Glymphatisches Systems entweder nicht oder in geringerem Ausmaße im Hirngewebe angereichert und vielleicht erst gar nicht einen bestimmten Schwellenwert überschreiten. Der Entsorgungsmechanismus würde damit auch die unzureichende Tätigkeit einer allzu durchlässigen Blut-Hirn-Schranke zumindest teilweise wieder korrigieren.
Die Existenz eines Glymphatischen Systems erklärte einmal mehr, warum ausreichender Schlaf für die Gesunderhaltung des Nervensystems wichtig ist.
Exogene Gift- und Schadstoffe als (Mit-)Verursacher einer Affektstörung aus kausaltheoretischer Sicht
Die Gefahr eines Ausbruchs oder der Verstärkung von Affektstörungen besteht immer dann, wenn Gift- oder Schadstoffe direkt oder indirekt zu Nervenfunktions- oder Gewebeschäden in den für die Entstehung und Verarbeitung von Emotionen zuständigen Hirnarealen führen. Abbildung 45 ist daher wiederum fast identisch mit den Graphiken vorhergehender Abschnitte.
ABBILDUNG 45: GIFT- UND SCHADSTOFFGEFÄHRDETE AREALE BEI AFFEKTSTÖRUNGEN
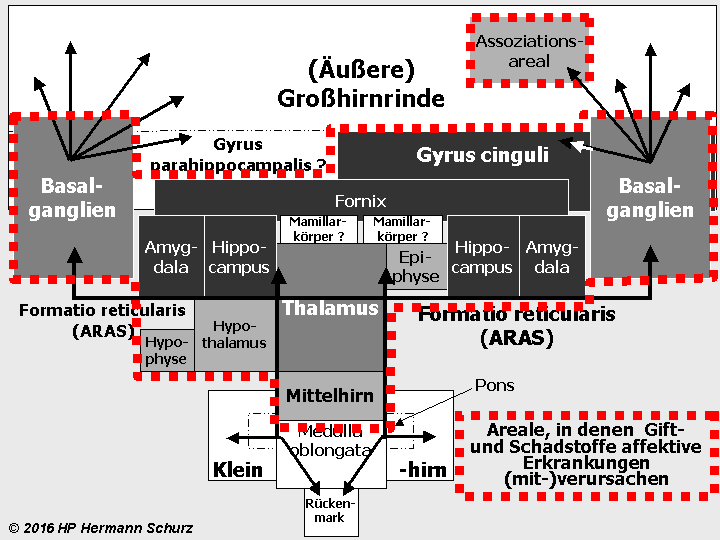
Abbildung 45: Alle markierten Hirnbereiche dienen der Steuerung und Regulierung von Emotionen und Affekten. Hier haben Gift- und Schadstoffe zwangsläufig erhebliche Auswirkungen und führen über funktionelle und/oder strukturelle Nerven- und Gliazellenstörungen bzw. Gewebe- oder vaskuläre Schädigungen potentiell zur Entstehung einer Affektstörung oder anderer neurologischer Erkrankungen. Es hat keine Bedeutung, ob Schäden durch die Substanzen direkt oder indirekt verursacht werden.
Empirische Erkenntnisse neurologisch-psychiatrischer Auswirkungen von Industriegiften
Anhand des Beispiels der hochgiftigen polychlorierten Biphenyle (PCB) lassen sich empirisch nachgewiesene neurotoxische Wirkungsmechanismen einer wichtigen Schadstoffgruppe gut nachvollziehen.
Die PCB-Gruppe wurde als Teil von 12 Substanzen/-gruppen gleich zum Start der Stockholmer POP-Konvention im Dezember 2000 verboten und zählt zu den damals als „Dreckiges Dutzend“ bezeichneten Schadstoffen. In vielen Ländern, so auch in der Bundesrepublik Deutschland, waren Herstellung und Verwendung von PCB allerdings schon vorher stark eingeschränkt oder nicht erlaubt.
Trotz ihres Verbots werden PCB-Verbindungen aufgrund ihrer Stabilität aktuell und in Zukunft ein Problem darstellen. Man verwendete sie in der Energie- und Elektrowirtschaft (Transformatoren, Kondensatoren), als Hydraulikflüssigkeit und in der Chemie- und Kunststoffindustrie als Weichmacher bis weit in die 1980er Jahre. PCB sind überall im Wasser, im Boden und in der Luft messbar. Auch in Deutschland sind viele Gebäude und Regionen nach wie vor PCB-belastet.
Bei PCB handelt es sich um eine Gruppe von 209 organischen Chlorverbindungen, deren akute Toxizität vergleichsweise gering ist, die jedoch schon bei geringen Mengen langfristig zu Vergiftungen führen. Sie lösen durch DNA-Veränderungen Krebs aus, verändern die Wirkung vieler Peptide, schädigen die Leibesfrucht, führen zu Organveränderungen und vor allem auch zu erheblichen Schädigungen des Nervensystems, denn sie wirken direkt und indirekt neurotoxisch. Beim Abbau von PCB im Körper entstehen häufig Abbau-Toxine, die noch giftiger sind als deren Ausgangssubstanzen.
Die direkte Neurotoxizität von PCB ist mit verschiedenen physiologischen Mechanismen zu begründen. Zunächst wirken PCB-Verbindungen unmittelbar auf Nervenzellen und führen dort zu Prozessstörungen, beispielsweise bei der Botenstoffsynthese. Ein weiterer Grund ist deren Eigenschaft, sich im Fettgewebe anzureichern. Da sowohl die Nerven- als auch wichtige Gliazellen einen hohen Fettanteil haben, ist das Gehirn besonders PCB-gefährdet. Die PCB-Konzentration in den fettreichen Membranen der Hirnzellen führt zu einer Störung der Membranrezeptoren. Folgen sind u. a. Störungen des zellulären Stoffaustauschs und Störungen der Nervenreizverarbeitung.
Beispiele indirekter neurotoxischer Wirkungen von PCB betreffen Leber und Schilddrüse. PCB sind lebertoxisch und durch die abhnehmende Fähigkeit der Leber, Giftstoffe zu neutralisieren, kommt es langfristig zu einer weiteren Anreicherung von PCB und anderer Schadstoffe im Gehirn. Auch die Schilddrüse wird durch PCB geschädigt. Das kann zu einer Schilddrüsenunterfunktion mit entsprechend negativen Folgen für das Gehirn führen.
PCB gelangen über Nahrung und Atmung in den Körper. Es ist nicht ganz klar, welcher Aufnahmepfad bei PCB die größere Rolle spielt.
Nahrungsgifte gelangen über den Darm ins Blut und über die Pfortader zunächst in die Leber und werden dort bei ausreichender Leberfunktion zumindest teilweise neutralisiert. Auch eine funktionierende Blut-Hirn-Schranke vermag den Giftstoffen im Blut den Zugang zum Gehirn zu verwehren. Beide Voraussetzungen sind jedoch häufig nicht erfüllt.
Eingeatmete PCB gelangen - neben der Aufnahme über die Lunge ins Blut - auch durch die Riechnerven unmittelbar ins Gehirn. Diese Nerven führen durch gefühls- und affektverarbeitende Hirnareale oder enden dort. Das betrifft beispielsweise die Amygdala oder den Hippocampus, darüber hinaus gibt es auch eine Verbindung in den olfaktorischen Cortex. Auf diese Weise können PCB emotionsverarbeitende Hirnstrukturen direkt erreichen und schädigen (Quelle: R. Apfelbach, A. Engelhart et al., The olfactory system as a portal of entry for airborne poychlorinated biphenyls (PCBs) to the brain?, Archives of Toxicolgy, Vol. 72 (5), 4/1998, Leibniz-Institut für Arbeitsforschung, Dortmund, Springer Verlag Heidelberg, Heidelberg 1998, https://link.springer.com/...).
Aufgrund dieses direkten Aufnahmemechanismus' via Riechzentrum ergibt sich die höchst beunruhigende Tatsache, dass im Blut gemessene PCB-Werte nicht unbedingt darauf rückschließen lassen, in welcher Höhe das Gehirn PCB-belastet ist. Fazit: Die über Luft und Nase aufnommenen Gifte erweisen sich zumindest für das Gehirn als mindestens genauso gefährlich wie diejenigen, die das Nervensystem über das Blut erreichen.
Nachfolgend einige Beispiele für die Neurotoxizität von PCB, die in Workshop Papers eines 1993 erschienenen Reports des Risk Assessment Forums der US-amerikanischen EPA-Behörde und Carole Braverman genannt wurden (Quelle: Carole Braverman, General Toxicity of PCBs, in: Workshop Report on Developmental Neurotoxic Effects Associated with Exposure to PCBs, 9/1992, Research Triangle Park, North Carolina/USA, Risk Assessment Forum, EPA U. S. Environmental Protection Agency, Washington, D. C./USA 1992, http://nepis.epa.gov/...):
- Das sich entwickelnde Gehirn von Kindern und Jugendlichen (→ Abschnitt 4.12.4) reagiert sehr empfindlich auf PCB, teilweise schon bei Mengen, die für Erwachsene keine Gefahr darstellen. Es ergaben sich Aufmerksamkeitsdefizite, Hyperaktivität, Entwicklungsstörungen, Minderungen des Lern- und Intelligenzvermögens, Verhaltensstörungen, beispielsweise aggressives Verhalten, und auch Störungen der Affekte (Quellen: Ted Schettler, Toxic Threats to Neurologic development of children, Environmental Health Perspectives, 109, Supplement 6, 12/2001, National Insititute of Environmental Health Sciences, Research Triangle Park, North Carolina/USA, 2001, https://www.jstor.org/...).
- Mehrere Studien belegen einen negativen Effekt von PCB auf den Dopaminstoffwechsel.
1991 ergab eine Zellstudie den Rückgang der Dopamin-Synthese nach PCB-Belastung (Quelle: W. Shain et al., Neurotoxicity of polychlorinated biphenyls: Structure-activity relationship of individual congeners, Toxicology and Applied Pharmacology, 10/1991 (111/1), Elsevier/U. K., https://www.sciencedirect.com/...).
Eine im selben Jahr veröffentlichte Tierversuchsstudie mit Makakenaffen und zwei PCB-Verbindungen ergab einen Rückgang der Dopaminkonzentrationen im dopaminergen System. Die Tiere erhielten über 20 Wochen zwei PCB-Verbindungen (Aroclor 1016 oder 1260) in drei verschiedenen Dosen. Ein Rückgang der Dopamin-Synthese wurde in den Basalganglien (Nucleus caudatus und Putamen, nicht jedoch im Pallidum) sowie im Hypothalamus festgestellt. Im Hippocampus konnte man keine Veränderungen erkennen. Eine der beiden PCB-Verbindungen reduzierte den Dopamingehalt in der Substantia nigra des Mittelhirns (Quelle: R. Seegal et al., Comparison of the effects of Aroclor 1016 and 1260 on non-human primate catecholamine function, Toxicology, 66, 1991, Elsevier Scientific Publishers Ireland Ltd., https://www.sciencedirect.com/...).
Eine weitere Studie aus dem Jahre 1993, ebenfalls unter der Leitung von Richard Seegal, kam zu vergleichbaren Ergebnissen und zeigte darüber hinaus, dass sich die Dopamin-Synthese auch nach Beendigung der PCB-Exposition nicht erholte (Quelle: R. Seegal et al., Decreases in dopamine concentrations in adult, non-human primate brain persis following removal from polychlorinated biphenyls, Toxicology, 86, 1994, Elsevier Scientific Publishers Ireland Ltd., http://www.sciencedirect.com/...).
In einer Untersuchung des Umweltbundesamtes aus dem Jahre 2012 wurden u. a. die Ergebnisse zur Neurotoxizität von PCB mehrerer Studien miteinander verglichen (Quelle: Ableitung von Human-Biomonitoring-(HBM)-Werten für Polychlorierte Biphenyle (PCB) im Blut, Bekanntmachung des Umweltbundesamtes und Stellungnahme der Kommission Human-Biomonitoring des Umweltbundesamtes, Dessau-Roßlau 2012, https://www.umweltbundesamt.de/...).
Es wurden 15 Studien mit Geburtskohorten aus Japan, den USA, Kanada, Grönland, Spanien, Deutschland, den Niederlanden, der Slowakei und von den Färöer-Inseln analysiert, die neurotoxische Effekte einer prä- bzw. postnatalen PCB-Exposition zum Gegenstand hatten. Nicht alle Studien ergaben signifikante neurotoxische Effekte, jedoch konnten in ihrer Mehrzahl niedrigere Leistungen der Kinder in neurologischen und neuropsychologischen Tests in Abhängigkeit von einer PCB-Exposition festgestellt werden.
Anzumerken ist, dass die PCB-Belastung bei allen Geburtskohorten aufgrund der Blutwerte verifziert wurde, zum Teil zusätzlich auch über die Muttermilch. Dabei wird die Möglichkeit unterschlagen, dass die Schadstoffe postnatal auch über andere Möglichkeiten verfügen, in das Gehirn zu gelangen, zum Beispiel über die Geruchsnerven. Eine mögliche direkte Belastung des Gehirns über das Riechzentrum lässt sich anhand von Blutwerten leider nicht erkennen.
Studien mit Untersuchungen an Erwachsenen, so stellten die Autoren der Publikation fest, ergaben ebenfalls häufig den Verdacht der Neurotoxizität von PCB, die Ergebnisse waren jedoch seltener und weniger spezifisch (→ Tabelle 24). Auch hier wurde nirgendwo die Möglichkeit einer direkten Intoxikation über das Riechzentrum berücksichtigt, so dass die PCB-entlastenden Schlussfolgerungen mit Vorsicht zu interpretieren sind.
TABELLE 24: ERWACHSENENSTUDIEN ZUR PCB-PROBLEMATIK IM VERGLEICH
Studie |
Neurotoxische bzw. neuropsychiatrische Effekte von PCB |
|---|---|
| Fischbein et al. 1979 (Arbeitsplatzstudie) | Konzentrationsprobleme, Kopfschmerzen, Schwindel, Depression, Müdigkeit, Beeinträchtigung der Merkfähigkeit, Schlafstörungen, Nervosität. Keine neurologischen Störungen nachgewiesen, jedoch keine differenzierten Testverfahren eingesetzt. |
| Emmett et al. 1988 (Arbeitsplatzstudie) | Kopfschmerzen, Schlafstörungen, Beeinträchtigungen der Merkfähigkeit. Autoren führten die Symptome nicht auf die PCB-Belastung zurück. |
| Broding et al. 2008 (Arbeitsplatzstudie) | Diverse Beschwerden wurden geäußert. Keine Korrelation mit den PCB-Belastungen im Blut. |
| Peper et al. 2005 (Arbeitsplatzstudie) | Diverse Beschwerden wurden geäußert. Reduzierte Leistungen in den Aufmerksamkeitstests, bessere Leistungen bei der Merkfähigkeit. Es konnte kein signifikanter Zusammenhang zwischen Symptomatik und PCB-Belastung festgestellt werden. |
| Singer 1988 (Arbeitsplatzstudie) | Diverse Symptome potentiell neurotoxischen Ursprungs wurden genannt. Es konnte eine Verminderung der Nervenleitgeschwindigkeit bei PCB-belasteten Teilnehmern festgestellt werden. |
| Troster et al. 1991 (Arbeitsplatzuntersuchung) | Kognitive Defizite und emotionale Irritationen bei zwei PCB-exponierten Personen. Ein Teilnehmer zeigte Anzeichen einer Demenz. Keine Veränderungen in der CT oder im EEG feststellbar. |
| Corrigan et al. 1998 (Post-Mortem-Studie) | Höhere PCB-Konzentrationen in den Hirngeweben Verstorbener, die an Morbus Parkinson erkrankt waren. |
| Seegal et al. 2004 | Gehäuftes Auftreten von Morbus Parkinson bei PCB-exponierten Arbeitern. |
| Steenland et al. 2006 | Leicht gehäuftes Auftreten von Morbus Parkinson und Demenz bei hoch PCB-exponierten Frauen. Ebenfalls mehr ALS-Erkrankungen bei PCB-exponierten Frauen. Signifikante Zusammenhänge zwischen Morbus Parkinson und einer PCB-Exposition konnten bezogen auf alle Teilnehmer nicht nachgewiesen werden. |
Tabelle 24: Das Umweltbundesamt verglich in ihrer Publikation u. a. die Ergebnisse verschiedener Studien mit Erwachsenen, die sich mit dem Aufspüren von Zusammenhängen zwischen einer PCB-Exposition und psychiatrisch-neurologischen Symptomen bzw. Erkrankungen beschäftigten (Quelle wie oben). Leider können potentielle Intoxikationen über die Riechnerven nicht gemessen werden, was insbesondere die PCB-entlastenden Interpretationen einiger Studienergebnisse infrage stellen.
Die Bekanntmachung des Umweltbundesamtes umfasst noch wesentlich mehr Studien mit Hinweisen auf nervenschädigende Wirkungen von PCB, auf dessen Darstellung hier aber verzichtet wird.
Im Fazit urteilten die Autoren: „Insgesamt besteht eine überzeugende Evidenzlage für adverse PCB-Effekte auf das sich entwickelnde Nervensystem. In den Geburtskohorten zeigten sich trotz der Heterogenität der Studiendesigns spezifische neurotoxische Effekte abhängig von der PCB-Belastung. (...) Studien an Erwachsenen liegen in deutlich geringerer Zahl vor und vermitteln kein klares Bild. Aus den zur Verfügung stehenden Daten entsteht der Eindruck, dass sich das Nervensystem von Erwachsenen als deutlich weniger anfällig gegenüber PCB-Belastungen erweist. Allerdings lässt die Mehrzahl der zur Verfügung stehenden Studien Mängel in der Expositionserfassung oder andere methodischen Schwächen erkennen, die eine Interpretation der Ergebnisse erschwert.“ (Quelle: wie oben).
Eine gute Übersicht über verschiedene Untersuchungen zur Neurotoxizität von Chlorverindungen (auch PCB) liefert eine Literaturstudie von Greenpeace UK aus dem Jahre 1995, der sehr übersichtlich wichtige Fakten zusammenfassend darstellt (Quelle: M. Allsopp, P. Johnston, P. Costner, Body of evidence - The effects of chlorine on human health, Greenpeace UK/London, 5/1995).
Erfreulicherweise hat Greenpeace Hamburg dazu einen Beitrag in deutscher Sprache verfasst. In den folgenden sieben Absätzen werden daraus Erkenntnisse über verschiedene schädliche Chlorverbindungen zitiert, darunter auch PCB (Quelle: W. Schwarz et al., Chlor macht krank - Die Auswirkungen von Chlorverbindungen auf die menschliche Gesundheit, Greenpeace e. V., 6/1995, Hamburg 1995, http://www.oekorecherche.de/...). Zu beachten ist, dass einige der damals gemachten Verwendungsangaben nicht mehr dem aktuellen Stand entsprechen, da die Übersichtsstudie 1995 erstellt wurde. Alle inhaltlichen Aussagen sind nach wie vor gültig.
Über die Neurotoxizität von PCB bzw. Dioxinen heißt es: „PCBs sind für das Nervensystem des Erwachsenen toxisch. (...) Untersuchungen bei Menschen wurden an Arbeitern durchgeführt, die nach Bränden von Transformatoren oder Kondensatoren PCBs und Dioxinen (PCDD/PCDFs) ausgesetzt waren (siehe Rogan und Gladen 1992). Zwei Studien zeigen, daß nach der Exposition Nervenschäden auftraten (distale Axonopathie). Eine andere Studie zu Feuerwehrleuten, die bei Löscharbeiten in einem Kraftwerk diesen Chemikalien ausgesetzt gewesen waren, zeigte Gedächtnischwäche und niedrigere Punktwerte bei kognitiven (Intelligenz-)Tests. Bei den Unfällen in Yusho und Yu-Cheng, wo PCB- und dioxinverseuchter Reis verzehrt wurde, fanden sich Anzeichen von Nervenschäden bei einigen exponierten Erwachsenen. Die Nervenschäden waren durch Verlangsamung der Übertragung von Nervensignalen in das periphere Nervensystem gekennzeichnet (Seegal und Shain 1992). Nicht immer werden schwerflüchtige chlororganische Verbindungen, die wegen ihres niedrigen Dampfdrucks gewöhnlich nicht zu hohen Luftkonzentrationen führen, auf dem Nahrungsweg aufgenommen. Besonders in Innenräumen können sich auch schwerflüchtige Verbindungen in der Atemluft so stark aufkonzentrieren, daß neurotoxische Effekte möglich sind, insbesondere dann, wenn Personen gegenüber solchen Stoffen langzeitexponiert sind. Das ist häufig in Wohnräumen oder Büros der Fall (Sick building syndrom etc.).“
Über Hexachlorcyclohexan (Technisches HCH) heißt es: „Belastende Luftkonzentrationen durch schwerflüchtige Verbindungen sind mitunter auch an industriellen Arbeitsplätzen anzutreffen. Während frühere Studien bei chronischer Niedrigdosis-Exposition keine Auswirkungen auf das Nervensystem gefunden hatten (Baumann et al. 1981), stieß eine neuere Studie zu 356 Arbeitern, die in einer indischen Fabrik HCH verarbeiteten, auf neurologische Symptome (Nigam et al. 1993). Die Belastung wurde durch Blutproben von HCH (a-HCH und b-HCH) bestimmt. Unter den Arbeitern wurden verschiedene Symptome gefunden, vor allem erhöhte Fälle von Kopfschmerzen, Schwindel und Übelkeit. Auch fanden sich bei einigen Arbeitern EEG-Abnormalitäten, die Störungen in den Gehirnfunktionen anzeigen. Diese neurologischen Symptome wurden mit der Intensität der HCH-Exposition der Arbeiter in Beziehung gesetzt und die HCH-Exposition als offenkundige Ursache erkannt.“
Über Pentachlorphenol (PCP) und γ-Hexachlorcyclohexan (Lindan) in Holzschutzmitteln heißt es: „Jene Symptome, nämlich eine Anhäufung relativ unspezifischer subjektiver Befindlichkeitsstörungen, wurden auch beim deutschen Holzschutzmittel-Skandal als Hauptwirkung von Langzeitexposition im Niedrigdosisbereich durch PCP und Lindan diagnostiziert. Sie wurden auch als 'Holzschutzmittelsyndrom' bezeichnet. Nach einer Schadensdokumentation der 'Interessengemeinschaft Holzschutzmittel-Geschädigter' haben sich mindestens 10.000 Personen in ihren Wohnungen durch das Einatmen langsam ausgasender Holzschutzmittel, die sie zum Teil selber in den siebziger und achtziger Jahren verstrichen hatten, in dieser Art vergiftet. Von betroffenen Patienten wurden in einer Stichprobe (Jüdt-Duve/Duve 1993, S. 85 ff.) am häufigsten Konzentationsstörungen, depressive Stimmungslage, Schlafstörungen, Merkfähigkeitsstörungen, Schwitzen und Ermüdbarkeit genannt. Diese Befunde decken sich mit einer Fall-Kontrollstudie an der Universitäts-Frauenklinik Heidelberg, bei der 15 Holzschutzmittel-Exponierte mit einem Mittelwert von 48,3 Mikrogramm PCP im Liter Blutserum und einer mittleren Expositionsdauer von 11 Jahren einer gleichgroßen Kontrollgruppe ohne Belastung gegenübergestellt wurden (Ertl o. J., S. 30). Es wurden folgende Vergleichswerte zwischen beiden Gruppen festgestellt: Erschöpfung (87% gegenüber 15%), Müdigkeit (87% gegenüber 33%), Schlafstörungen (80% gegenüber 15%), Energielosigkeit (67% gegenüber 25%), Libidostörungen (66% zu 0%), Reizbarkeit (60% zu 31%), Antriebslosigkeit (13% zu 0 %). Der Hamburger Mediziner Fabig (1992) dignostizierte bei Holzschutzmittel-Geschädigten mithilfe eines radiologischen Untersuchungsverfahrens eine Veringerung des 'regionalen cerebralen Blutflusses', die er mit der Belastung durch PCP, Lindan, Dioxinen und organischen Lösemitteln in Verbindung brachte. Veränderte regionale Hirnstromaktivität stellte der Schleswiger Neurologe Lohmann (1989) mithilfe einer EEG-Weiterentwicklung (brain mapping) fest. Außer Befindlichkeitsstörungen (Leistungsminderung, Konzentrationsschwäche usw.) diagnostizierten Huber et al. (1990) auch Immunstörungen bei pathologisch belasteten Probanden (PCP-Wert im Blut über 20 Mikrogramm/Liter). Dies liegt aufgrund der Wechselwirkungen zwischen Nervensystem und Immunsystem nahe (Reichert 1990, S. 260).“
Über Perchlorethylen (PER) heißt es: „Neben etwa 5.000 Tonnen für Chemische Reinigungen geht heute die Hauptmasse der inländischen Anwendung von Perchlorethylen (PER) in die Metallindustrie zur Metallentfettung. Ca. 20.000 Tonnen PER werden jährlich als Ersatz für verflüchtigtes Lösemittel eingesetzt bzw. emittiert. Nachdem ein teilweiser Umstellungsprozeß auf wäßrige Verfahren mittlerweile weitgehend abgeschlossen ist, dürfte sich der Jahresbedarf auf diesem Niveau einpendeln, wobei die verbleibende Anwendung (Feinstreinigung) tendenziell nur noch in emissionsarmen Anlagen geschieht (Leisewitz/Schwarz 1994). Weder solche modernen Anlagen noch die Einhaltung der zulässigen MAK- und BAT-Werte bieten jedoch für die Arbeitsplätze in der näheren betrieblichen Umgebung absolute Sicherheit vor PER-bedingten Beschwerden, wie ein Fallbericht aus einer süddeutschen Fabrik für elektronische Schaltungen zeigt (Seeger 1991). Trotz Unterschreitung des MAK-Wertes von 50 ppm (= 345 mg/m3) durch Meßwerte zwischen, je nach Abstand von der Waschanlage, 5 und 15 ppm klagten die Arbeiter in der Stanzerei immer wieder über Kopfschmerzen, Rötung der Augen und der Haut, Probleme mit den Atemwegen, vereinzelt Nasenbluten, Magenbeschwerden und Müdigkeit. Auch der BAT-Wert, der bei 1.000 Mikrogramm pro Liter Blut lag, wurde nicht überschritten: Während die Referenzbelastung der Gesamtbevölkerung unter 1 Mikrogramm im Liter Blut betrug, die bei Beschäftigen in anderen Produktionsräumen des gleichen Betriebs (0,5 Mikrogramm/l) auch gemessen wurde, fanden sich bei den über neurologische Symptome klagenden Arbeitern im Umfeld von 15 Metern Entfernung von der neuen PER-Reinigungsanlage Konzentrationswerte zwischen 400 und 720 Mikrogramm/Liter. Daß es sich bei den in Metallbetrieben mit PER-Reinigungsanlagen auftretenden subjektiven Befindlichkeitsstörungen um typische neurotoxische Effekte des Lösemittels Perchlorethylen handelt, belegte eine Untersuchung von Böttger (1989) an 45 Chemisch-Reinigern. Die Studie entstand im Zusammenhang mit der seit 1987 in der Öffentlichkeit geführten Diskussion um Wohnraumbelastungen in der Nachbarschaft Chemischer Reinigungen. (Bis 1990 waren bei über 2.000 Messungen in verschiedenen deutschen Großstädten in Abhängigkeit von der Entfernung zum Reinigungsraum Blutwerte bei der Wohnbevölkerung bis zu 100 Mikrogramm PER pro Liter und PER-Konzentrationen in fetthaltigen Lebensmitteln bis zu 100 Milligramm pro Kilogramm - u. a. in Butter und Speiseeis - gemessen worden). Die von Böttger zusammengestellte Gruppe von 45 Chemisch-Reinigern aus Düsseldorf klagte signifikant häufiger über gesundheitliche Beschwerden als eine durch PER nicht belastete Referenzgruppe von 106 Personen. Das galt u. a. für abnorme Müdigkeit, Gedächtnisstörungen, Konzentrationsschwäche, Nervosität, Sprachstörungen, Schweißausbrüche, Völlegefühl. Zugleich wurden innerhalb der Reinigergruppe auf der Basis regelmäßiger Blutmessungen signifikante Unterschiede in der Klagehäufigkeit von Reinigern mit höheren PER-Blutwerten gegenüber geringer blutbelasteten Reinigern deutlich. Die Blutkonzentrationen der 45 Reiniger stieg übrigens von montags bis freitags kontinuierlich an (von ca. 150 Mikrogramm/l auf 415 Mikrogramm/l). Die immer noch hohen Eingangsblutwerte am folgenden Montag zeigten, daß ein expositionsfreies Wochenende nicht ausreicht, um das aufgenommene PER in ausreichendem Maß aus dem Körper wieder zu entfernen. Der Vorsorgerichtwert pro Kubikmeter Innenraumluft ist 1990 für Wohnungen auf 0,1 Milligramm pro Kubikmeter gesenkt worden (der MAK-Wert blieb bei 345 Milligramm pro Kubikmeter!), und für Lebensmittel wurde ein PER-Höchstgehalt von 0,1 Milligramm pro Kilogramm festgelegt (Kommunale Briefe für Ökologie 22/1990). Die Immissionsschutzauflagen für Chemisch-Reinigungen wurden inzwischen zwar drastisch verschärft. Dennoch zeigten sich die Gesundheitsexperten der Stadt Frankfurt am Main nach einer Kontrolluntersuchung 1992 skeptisch, ob durch verbesserte Reinigungstechnik die PER-Belastung der Anwohner ausreichend gesenkt werden könne, weil auch die Neuanlagen zu 81% die zulässigen Immissionswerte überschritten (Stadt Frankfurt 1992).“
Über Trichlorethen (TRI) heißt es: „Die Toxizität von TRI, das heute in der Metallentfettung und in Gummiklebstoffen (Schwarz/Leisewitz 1994) im Umfang von etwa 10.000 Tonnen jährlich eingesetzt wird, beruht auf den physiologischen Wirkungen auf das Zentralnervensystem, hauptsächlich in der Depression der Funktionen des ZNS nach akuten toxischen Einwirkungen. Wie 1,1,1-Trichlorethan wird es von Schnüfflern gern eingeatmet, weil es einen euphorischen Zustand, der zur Sucht führen kann, erzeugt. Barett et al. (1984) fanden einschlägige Symptome wie Benommenheit, Kopfschmerz und Müdigkeit bei einer Untersuchung von 188 Arbeitern mit TRI-Exposition (Durchschnittsalter 41 Jahre, durchschnittliche Expositionsdauer 7 Jahre). Die Befunde waren sämtlich statistisch signifikant. Eine epidemiologische Studie von Bowler et al. (1991) konstatierte bei 180 ehemaligen Arbeiterinnen der Mikroelektronikindustrie gegenüber einer Vergleichsgruppe signifikant geringere Leistungen bei Aufmerksamkeit, Konzentration, kognitiver Flexibilität, Gedächtnisfunktion und visuo-motorischer Beweglichkeit. Trichlorethylen schädigt das Axon (die Verbindungsleitung) der Nervenzellen, im Sinne eines 'chemischen Abschneidens' der Verbindung (Eisenbrand 1994, 85).“
Über 1,1,1-Trichlorethan heißt es: „Das ozonschichtschädigende, aber angeblich humantoxisch harmlose Lösemittel (z. B. in Tippex-Verdünner) war vor seinem Verbot (1990) als leichtes Rauschmittel beliebt, weil es in höheren Dosen narkotische und Rausch-Wirkung entfaltet. Drei Todesfälle innerhalb 6 Monaten in Schottland (Mac Dougall et al. 1987) haben zu vermehrter Vorsicht gegenüber diesem Lösemittel geführt (Frentzel-Beyme/Domizlaff, S. 155).“
Über Methylenchlorid (Dichlormethan) heißt es: „Das unter Krebsverdacht stehende Methylenchlorid wurde Anfang der 90er Jahre im Umfang von weit über 10.000 Tonnen in offenen Anlagen und Anwendungsformen eingesetzt: bei der professionellen Metall- und Holzentlackung, als Abbeizmittel für Lacke im Heimwerkerbereich, als Löse- und Treibmittel in Insektensprays oder Sprays für Kfz-Werkstätten und (mit 700 Tonnen) bei der Folienbeschichtung von PVC-Fenstern (Schwarz/Leisewitz 1994). Zentralnervöse Depression (Benommenheit, Müdigkeit, Augenirritation, Reizhusten) traten bei einem Schreiner auf, der 2 1/2 Jahre lang Methylenchlorid zum Abbeizen von Möbeln verwendet hatte (Shusterman et al. 1990). Arbeitsplatzmessungen ergaben eine Konzentration von 350 ppm, der Schreiner trug allerdings Schutzbrille und Atemgerät. Die Symptome ließen jeweils an Wochenenden und im Urlaub nach. Nach Verbesserung der Atemschutzeinrichtung (häufigeres Wechseln der Gaspatronen) fielen die Symptome weg. Bedeutsam sind die Befunde einer Kopplung von Kohlenmonoxid aus dem oxidativen Abbau von Methylenchlorid an Hämoglobin (Frentzel-Beyme/Domizlaff, S. 35). Letzteres stimmt mit früheren in-vitro-Studien (Ahmed et al. 1977) überein, denen zufolge die Bioatransformation des Methylenchlorids zu Kohlenmonoxid den Sauerstofftransport des Blutes behindert und somit eine akut-toxische Wirkung hervorruft. Daher ist für Methylenchlorid ein BAT-Wert von 5% Carboxyhämoglobin im Blut festgelegt (DFG 1994a, S. 143).“
Bei den Schadstoffen Trichlorethen (TIR) und Perchlorethylen (PER) konnten Zusammenhänge mit einem Rückgang der Dopamin-Synthese bzw. einer Erkrankung an Morbus Parkinson nachgewiesen werden. In einer internationalen Studie aus dem Jahre 2011 unter Leitung von Samuel Goldmann, Mitarbeiter des kalifornischen Parkinson's Institute and Clinical Center in Sunnyvale/USA, wurden 99 Zwillingspaare mit jeweils einem an Morbus Parkinson erkrankten Zwilling untersucht (Quelle: S. M. Goldman et al., Solvent Exposure and Parkinson Disease Risk in Twins, Annals of Neurology, 6/2012, Wiley-Blackwell, Hoboken/New Jersey, USA 2012, http://onlinelibrary.wiley.com/...).
Untersucht wurde, ob eine Exposition mit sechs Lösungsmitteln die Entstehung von Morbus Parkinson begünstigen, darunter Trichlorethen und Perchlorethylen. Die Forscher berechneten ein sechsfach höheres Erkrankungsrisiko bei Trichlorethen. Bei der Betrachtung beider Lösungsmittel gemeinsam ergab sich sogar ein neunfach höheres Risiko, an Morbus Parkinson zu erkranken. Ebenfalls gab es einen Zusammenhang zwischen der Expositionsdauer und der Erkrankung. Bei den restlichen vier Chemikalien wurde kein erhöhtes Riskio festgestellt.
Empirische Erkenntnisse neurologisch-psychiatrischer Auswirkungen von Staubpartikeln und NO2
Über zentralnervöse Wirkungen mikroskopisch kleiner Luftpartikel, dem sogenannten Feinstaub, weiß man bisher nur wenig. Die meisten Erkenntnisse betreffen Auswirkungen auf Lungenfunktion und das Herz-Kreislauf-System. Erste Studien deuten auf mögliche negative Folgen direkten und indirekten Ursprungs auch für das Gehirn.
Unter dem Feinstoffbegriff werden drei Schadstoffgruppen verstanden, die sich nur durch ihre Größe unterscheiden. Feinstoff PM10 hat einen Durchmesser von maximal 10 µm, lungengängiger Feinststaub PM2.5 hat einen Durchmesser von weniger als 2,5 µm und dringt bis in die Alveolen der Lungen vor.
Die gefährlichen ultrafeinen Partikel sind im Durchmesser kleiner als 0,1 µm und gelangen aufgrund ihrer geringen Größe in den Blutkreislauf. Eine weitere potentielle Eintrittspforte ultrafeiner Partikel in den menschlichen Körper sind die Riechnerven, die sich auch bei eingeatmeten Industriegiften schon als direkter Zugang zum Nervensystem erwiesen, insbesondere in das Gehirn und die affektrelevanten Hirnbereiche. Mehrere Tierversuchsstudien an Ratten, Nagetieren und Affen bestätigten die Vermutung und es gibt Hinweise darauf, dass es in der Folge zu entzündlichen Veränderungen im Gehirn kommen kann (Quellen: A. Elder, G. Oberdörster et al., Translocation of inhaled ultrafine manganese oxid particles to the central nervous system, Environmental Health Perspectives, 8/2006, National Insititute of Environmental Health Sciences, Research Triangle Park, North Carolina/USA, 2006, https://www.researchgate.net/publication/... und G. Oberdörster et al., Translocation of inhaled ultrafine particles to the brain, Inhalation Toxicology, 6/2004, Taylor & Francis Group, Milton Park/U. K. 2004, https://www.researchgate.net/publication/...).
Eine Studie aus dem Jahre 2014 unter Leitung von Elissa Wilker untersuchte die Auswirkungen einer Belastung mit lungengängigem Feinststaub (Durchmesser von weniger als 2,5 µm, aber nicht unter 0,1 µm) auf das Gehirn (Quelle: E. H. Wilker et al., Long-Term Exposure to Fine Particulate Matter, Residential Proximity to Major Roads and Measures of Brain Structure, Cardiovascular Epidemiology Research Unit, Department of Medicine, Beth Israel Deaconess Medical Center, Boston/MA, USA, Stroke Journal of the American Heart Association, 4/2015, John Wiley & Sons, Hoboken/New Jersey, USA 2015, https://www.ahajournals.org/...).
Studienziele waren die Bewertung möglicher Zusammenhänge zwischen der lungengängigen Feinststaubbelastung und dem neurovaskulären Hirnzustand bzw. dem Hirnvolumen. Die Untersuchungen erfolgten über MRT-Auswertungen der Gehirne 900 älterer und gesunder Probanden (> 60 Jahre), die in unterschiedlicher Entfernung zu Hauptverkehrsstraßen wohnten und damit unterschiedlichen Feinststaubbelastungen ausgesetzt waren. Es wurden die cerebralen Blutgefäße und die Blutversorgung untersucht einschließlich der Suche nach Anhaltspunkten für Blutgerinnsel und stille Hirninfarkte. Darüber hinaus erfolgte eine Vermessung verschiedener Hirnvolumina (u. a. Gesamthirn und Hippocampus).
Die Untersuchungen ergaben cerebrale Auswirkungen der Feinststaubexposition. So hatten höher belastete Probanden ein geringeres Hirnvolumen. Ebenfalls konnten Wilker und ihr Team ein um 46% erhöhtes Riskio für stille Hirninfarkte bei stark Feinstaubbelasteten nachweisen. Dazu Elissa Wilker: „Das ist besorgniserregend, denn wir wissen, dass stille Hirninfarkte sowohl das Apoplex-Risiko erhöhen als auch Demenz, Bewegungsstörungen und eine Depression zur Folge haben können.“ (Quelle: Long-Term Exposure to Air Pollution May Pose Risk to Brain Structure, Cognitive Functions, Pressemitteilung des Beth Israel Deaconess Medical Center, Harvard Medical School Teaching Hospital vom 23.4.2015, Boston/MA, USA, http://www.bidmc.org/...).
Die genauen Gründe der Neurotoxizität sind unklar. Auf jeden Fall kommen bei lungengängigem Feinststaub nur indirekte Zusammenhänge infrage, da dieser aufgrund seiner Größe weder in die Blutbahn gelangen kann noch in der Lage ist, über die Riechnerven das Gehirn direkt zu erreichen. Wilker und ihr Team vermuten daher, dass Partikel in den Lungenbläschen eine systemische Entzündung auslösen mit entsprechenden negativen Folgen für den gesamten Körper und damit auch für die Blutgefäße des Gehirns (Quelle: Pressemitteilung, wie oben).
Es ist anzumerken, dass parallele Auswirkungen ultrafeiner Partikel oder anderer Luftemissionen bei der Studie nicht ausgeschlossen werden können und es ist unklar, ob und wie diese Belastungen aus den statistischen Ergebnissen herausgerechnet wurden oder ob dies überhaupt möglich ist. Mit hoher Wahrscheinlichkeit korrelieren jedoch viele Luftschadstoffe in der gleichen Weise mit dem Wohnort ‑ insbesondere alle Arten von Feinstaubpartikeln ‑, so dass die Studienergebnisse eventuell auch auf andere Substanzen zurückzuführen sind. Das ändert allerdings nichts Grundsätzliches in der Bewertung der Folgen und Gefährlichkeit partikulärer Luftschadstoffe.
Ein Zusammenhang zwischen der Belastung mit Fein- und Feinststaub bzw. Feinststaub und Stickstoffemissionen und einem höheren Selbstmordrisiko wurde in zwei voneinander unabhängigen Studien in Südkorea und im US-Bundesstaat Utah festgestellt.
2010 veröffentlichen Chang Soo Kim und ein Team der südkoreanischen Yonsei University eine Studie über Zusammenhänge zwischen einer Belastung mit Feinstaub (PM10) bzw. Feinststaub (PM2.5) und suizidalem Verhalten während eines Zeitraums von einem Jahr (Quelle: C. Kim et al., Ambient particulate matter as a risk factor for suicide, Department of Preventive Medicine and Institute for Enviornmental Research, Yonsei University College of Medicine, Seoul, Republic of Korea, The American Journal of Psychiatry, 9/2010, 167 (9), American Psychiatric Association, Arlington/Virginia, USA, http://www.ncbi.nlm.nih.gov/pubmed/...).
Die Untersuchung wurde während des Jahres 2004 für PM10 an 106 Standorten in sieben südkoreanischen Großstädten und für PM2.5 an 13 Standorten in einer Stadt durchgeführt. Es wurden alle 4.341 Suizide in den sieben Städten während dieses Zeitraums berücksichtigt. Die Messung der Belastungswerte erfolgte stündlich. Verschiedene chronische Vorerkrankungen der Verstorbenen wurden recherchiert und berücksichtigt und zu Gruppen zusammengefasst. Sie betrafen Herz-Kreislauf-Erkrankungen, Diabetes mellitus, COPD, Krebserkrankungen und psychische Erkrankungen. Die Daten wurden um externe Faktoren, beispielsweise Wetter und Feiertagseinflüsse, bereinigt.
Laut Erhebungen stieg nach einer starken kurzfristigen PM10-Erhöhung über den Durchschnittswert die Wahrscheinlichkeit, einen Suizid innerhalb der folgenden zwei Tage zu begehen, über alle Gruppen um 9%.
Innerhalb der Gruppen von Suizidopfern mit verschiedenen Vorerkrankungen stiegen die Suizid-Wahrscheinlichkeiten bei Diabetes um 22%, bei Herz-Kreislauf-Kranken um 18,9%, bei COPD um 15,6% und unter Krebskranken um 10,5% und lagen auch über den Werten der Suizidopfer ohne die jeweiligen Erkrankungen.
Erstaunlicherweise erhöhte sich die Suizidwahrscheinlichkeit innerhalb der Gruppe der psychiatrisch Vorerkrankten nur um 8,1% und lag damit sogar leicht unterhalb des Durchschnittswertes von 9%. Innerhalb der Gruppe ohne psychiatrische Vorerkrankung lag die Steigerung mit 9,2% nur leicht über dem Durchschnittswert über alle Gruppen.
Die Ergebnisse für die PM2.5-Messungen wichen leicht ab, im Grundsatz war das Ergebnis ähnlich, obwohl die Situation nur in einer Großstadt untersucht wurde. Die Suizidwahrscheinlichkeit über alle Gruppen stieg hier um 10,1%.
Forscher der Universität Utah/USA unter der Leitung von Amanda Bakian untersuchten Zusammenhänge zwischen dem kurzfristigen Anstieg von Stickstoffdioxid, Schwefeldioxid, Feinstaub PM10 bzw. lungengängigem Feinststaub PM2.5 in der Luft und vollzogenen Selbstmorden im Landkreis Salt Lake County, der Teil des US-Bundesstaates Utah ist. Sie beriefen sich dabei auch auf die oben erwähnte Feinstaub-Studie aus Südkorea und vergleichbare Studien aus Taiwan und Kanada über andere Luftschadstoffe (Quelle: A. V. Bakian et al., Acute Air Pollution Exposure and Risk of Suicide Completion, Department of Psychiatry, School of Medicine, University of Utah, Salt Lake City, American Journal of Epedemiology, 2/2015, Oxford University Press Journals, Oxford/U. K. 2015, http://www.ncbi.nlm.nih.gov/...).
In der eher ländlich geprägten Region um die mit ca. 190.000 Einwohnern größten Stadt Salt Lake City ereigneten sich in den Jahren von 2000 bis 2010 insgesamt 1.546 Suizide bei durchschnittlich 964.000 Einwohnern des Landkreises Salt Lake County.
Die Analysen ergaben signifikante Anstiege der Zahl der Selbstmorde bei einer kurzfristig erhöhten Menge von Luftschadstoffen für die ersten drei Tage nach dem Ereignis. Bei PM2.5 erhöhte sich die Suizidwahrscheinlichkeit um 6%, bei Stickstoffdioxid sogar um 25%. Die stärksten Zusammenhänge ergaben sich dabei bei Männern und in der mittleren Altersgruppe der 36 bis 64jährigen und deuten damit auf unterschiedliche Vulnerabilitäten bei Geschlechtern und Altersgruppen hin.
Spezielle Untersuchungen über Wirkungen von Gift- und Schadstoffen auf Affekte
In einigen der bisher zitierten Arbeiten werden Zusammenhänge zwischen einer Gift- oder Schadstoffbelastung und Affektstörungen nur indirekt thematisiert. Es gibt leider nur wenige Studien, die sich direkt mit dieser Problematik auseinandersetzen.
Die Hirnforscher Laura Fonken und Randy J. Nelson studierten im Jahre 2011 mit einem Team der Universitiät von Ohio/USA mittels Tierversuchs die Auswirkungen schadstoffbelasteter Luft (Quelle: L. Fonken, R. J. Nelson et al., Air pollution impairs cognition, provokes depressive-like behaviors and alters hippocampal cytokine expression and morphology, Department of Neuroscience, The Ohio State University, Columbus, Ohio/USA 2011, Molecular Psychiatry, 16/2011, Nature Publishing Group, Macmillan Publishes Limited, London/New York/Tokio, http://www.ncbi.nlm.nih.gov/...).
Dazu wurden männliche Mäuse in zwei Gruppen geteilt. Die eine Gruppe erhielt für den Zeitraum von 10 Monaten zum Atmen schadstofffreie gefilterte Luft, die andere Gruppe bekam im selben Zeitraum mit Feinststaubpartikeln PM2.5 belastete Luft, was der Situation schadstoffbelasteter Großstädte entsprechen soll. Danach wurden verschiedene kognitiv-affektive und motorische Leistungen überprüft und die Gehirne der Tiere strukturell untersucht.
Die Verhaltensweisen der luftschadstoffbelasteten Tiere wiesen durchweg Merkmale auf, die mit denen einer Depression beim Menschen vergleichbar waren. Die Tiere schnitten bei den Lerntests schlecht ab, sie konnten sich einen bestimmten Weg durch ein Labyrinth schlecht merken und lösten diese Aufgaben mit deutlich weniger Erfolg als die schadstoffunbelastete Gruppe. In einem Schwimmtest gaben die schadstoffbelasteten Tiere frühzeitiger auf, in einem anderen Versuch zeigte diese Gruppe ängstliches Verhalten.
Die Untersuchung der Hirnstruktur der Tiere ergab Schädigungen der Nervenzellen im Hippokampus, vor allem degenerative Veränderungen der Dendriten, und man fand Hinweise auf Entzündungsreaktionen aufgrund vermehrter Anwesenheit von Botenstoffen des Immunsystems, beispielsweise TNF‑α.
Florian Rötzer, Herausgeber des Online-Magazins Telepolis, fasst die Aussagen der Fonken-Studie prägnant zusammen: „Kognitive Behinderungen und Depression würden, so die Wissenschaftler, miteinander einhergehen und seien verbunden mit Änderungen im Hippocampus. Das habe man bei Menschen und Nagetieren in anderen Studien erkennen können. (...) Aus ihrer Studie ergebe sich, dass eine anhaltende Aussetzung an Feinstaubkonzentrationen, wie sie für manche Städte besonders in den Dritten Welt oder in China und Indien typisch seien, zu «Neuroinflammation, Veränderungen im Hippocampus, Verhaltensveränderungen und abnehmenden kognitiven Fähigkeiten» bei Mäusen führen können. Zu vermuten ist, dass dies bei Menschen auch der Fall ist. Die Neurowissenschaftlerin und Lead-Autorin Laura Fonken fasst die Implikationen der Studie so zusammen: «Die Ergebnisse legen nahe, dass längere Aussetzung an verschmutzte Luft sichtbare negative Auswirkungen auf das Gehirn haben kann, die zu einer Reihe von gesundheitlichen Problemen führen können. Das kann wichtige und beunruhigende Implikationen für Menschen haben, die in städtischen, durch Luftverschmutzung belasteten Gebieten überall auf der Welt leben.»“ (Quelle: Florian Rötzer, Feinstaub kann zu Depressionen, Angst und kognitivem Leistungsabfall führen, Telepolis vom 6.7.2011, Heise Medien GmbH & Co. KG, Haar 2011).
Eine Studie des Instituts für Arbeitsmedizin und Sozialmedizin der Uniklinik RWTH Aachen aus dem Jahre 2014 unter Leitung von Petra Gaum und Thomas Kraus weist Zusammenhänge einer arbeitsbedingten Belastung mit PCB und psychiatrischen Erkrankungen nach (Quelle: P. Gaum, T Kraus et al., Prevalence and incidence rates of mental syndromes after occupational exposure to polychlorinated biphenyls, Institut für Arbeitsmedizin und Sozialmedizin der Uniklinik RWTH Aachen, International Journal of Hygiene and Environmental Health, Nr. 217/2014, Elsevier/Deutschland 2014, http://www.sciencedirect.com/...).
Untersucht wurden Zusammenhänge zwischen der Höhe der PCB-Belastung und der Schwere verschiedener Symptome somatoformer Störungen, von Depression, Angst- und Panikstörungen und der Häufigkeit von Symptomen bzw. dem Erkrankungsrisiko in einer Kohorte von 136 arbeitsbedingt PCB-belasteten Personen. Die PCB-Werte wurde im Blut bestimmt, die Erkrankungsymptome mit einem standardisierten Screening-Verfahren erfasst.
Die Ergebnisse der Kohorte wurden u. a. mit den Werten der durchschnittlich PCB-belasteten Bevölkerung in Deutschland verglichen. Bezüglich der Häufigkeit von Symptomen wurden niedrig- und hochbelastete Teilnehmer miteinander verglichen.
So waren die Neuerkrankungen (Inzidenzen) in Bezug auf alle Syndrome höher in den höher PCB-belasteten Gruppen. Unterschiede zeigten sich bei verschiedenen PCB-Verbindungen. So hatten höherchlorierte PCB (HPCB) und dioxinähnliche PCB (DIPCB) stärkere Wirkungen im Gegensatz zu niederchlorierten PCB (LPCB). LPCB haben eine deutlich niedrigere Halbwertzeit und das könnte ein Grund für die unterschiedichen Wirkungen sein.
Die Forscher vermuten einen Zusammenhang mit hormonellen Veränderungen durch Schädigungen der Schilddrüse oder Verminderungen der Neurotransmitter-Synthese und verweisen auch auf die weiter oben schon zitierten Studien, die Rückgänge der Dopamin-Synthese zeigten.
In der Zusammenfassung der Studienergebnisse schreiben die Autoren: „Zu allen 3 Messzeitpunkten (MPZ) zeigte die vorliegende Stichprobe, mit Ausnahme des somatoformen Syndroms, für alle Syndrome höhere Prävalenzen von psychischen Syndromen im Vergleich zur Normalbevölkerung (1). Zusätzlich fanden wir für die höherbelastete Gruppe höhere Prävalenzen als für die niedrigbelastete Gruppe (2) [z. B. für depressives Syndrom zu MZP 1: Höherbelastete 16% vs. Niedrigbelastete 1.2%]. Insgesamt haben die höher-PCB-belasteten Teilnehmer ein bis zu 15mal höheres Risiko, zukünftig ein psychisches Syndrom zu entwickeln als niedrig-belastete Personen (3). (...) Wir konnten zeigen, dass Personen mit einer erhöhten PCB-Belastung ein größeres Risiko für die Entwicklung eines psychischen Syndroms haben als Personen mit einer niedrigen PCB Belastung. Ein weiterer Schritt ist nun zu untersuchen, auf welche physiologischen oder psychologischen Prozesse diese Ergebnisse zurückzuführen sind, um gezielte Präventionsmaßnahmen einleiten zu können.“ (Quelle: Forum Arbeitsphysiologie - 17. Symposium Arbeitsmedizin und Arbeitswissenschaft für Nachwuchswissenschaftler, S. 22, Herausgeber B. Steinhilber, M. A. Rieger, Institut für Arbeitsmedizin, Sozialmedizin und Versorgungsforschung Tübingen, 11/2013).
Im September 2014 veröffentlichte die Zeitschrift Environmental Health Perspectives die Ergebnisse einer Langzeitstudie der Auswirkungen von Pestiziden bei (männlichen) Landwirten bzw. Landarbeitern in den USA, die sich über etwa zwölf Jahre erstreckte. Freya Kamel und ein Team von Wissenschaftlern der University of North Carolina at Chapel Hill in Zusammenarbeit mit den National Institutes of Health und dem National Institute of Environmental Health Sciences untersuchten Zusammenhänge zwischen der Verwendung verschiedener Pestizide und dem Auftreten einer Depression (Quelle: F. Kamel et al., Pesticide Exposure and Depression among Male Private Pesticide Applicators in the Agricultural Health Study, University of North Carolina at Chapel Hill, National Institutes of Health, Dept. of Health and Human Serivces, Environmental Health Perspectives, 122 (9), 9/2014, National Institute of Environmental Health Sciences, Research Triangle Park/North Carolina, USA 2014, http://ehp.niehs.nih.gov/...).
Mit den Auswirkungen landwirtschaftlich genutzter Giftstoffe auf das Zentralnervensystem beschäftigte sich Freya Kamel auch schon in einigen ihrer früheren Untersuchungen. Im Jahre 2006 wies sie Zusammenhänge zwischen der Anwendung von Pestiziden bzw. Herbiziden und Morbus Parkinson nach, beispielsweise für das Herbizit Paraquat (Quelle: F. Kamel et al., Pesticide Exposure and Self-reported Parkinson’s Disease in the Agricultural Health Study, American Journal of Epidemiology, 11/2006, 165 (4), Oxford University Press/Johns Hopkins Bloomberg School of Public Health, Baltimore/Maryland, USA 2006, http://aje.oxfordjournals.org/...). Im Jahre 2012 stellte Freya Kamel in einer anderen Studie Zusammenhänge zwischen Pestiziden und der Entstehung der Amyotrophen Lateralsklerose (ALS) fest, bei der sich Motorneurone im Gehirn und Rückenmark degenerativ verändern (Quelle: F. Kamel et al., Pesticide exposure and amyotrophic lateral sclerosis, Neurotoxicology 6/2012, 33, Elsevier/NL, 2012, http://www.sciencedirect.com/...).
So scheint es fast zwangsläufig, die Studiendaten auch auf potentielle Zusammenhänge zwischen der Pestizidanwendung und der Entstehung von Affektstörungen zu untersuchen. Darüber hinaus handelte es sich bei der aufwändigen Untersuchung eines Zeitraums von zwölf Jahren um ein „Leuchtturmprojekt“, das über seine Signalwirkung weitere Forschungen initiieren sollte.
Eine vorab durchgeführte Literaturrecherche verweist auf verschiedene Untersuchungen, die Zusammenhänge zwischen der Nutzung von Pestiziden und einer Depression feststellten.
In die Studie wurden 21.208 landwirtschaftliche Privatanwender (ausschließlich Männer) aus den beiden US-Bundesstaaten Iowa und North Carolina aufgenommen. Untersucht wurden 50 spezielle Pestizide aus zehn Pestizid-Gruppen. Die Teilnehmer wurden sukzessive in die Untersuchung einbezogen, die Starttermine lagen zwischen den Jahren 1993 und 1997, beendet wurden die Untersuchungen mit einem Follow-up per Telefonbefragung zwischen den Jahren 2005 und 2010. Es wurden vier Teilnehmergruppen gebildet in Abhängigkeit von der Frage, zu welchem Zeitpunkt die Diagnose einer Depression erfolgte: während des Studienverlaufs, nur zu Beginn der Studie, zu Beginn und bei Studienende oder nur bei Studienende.
Es ergab sich ein signifikanter Zusammenhang zweier Pestizid-Gruppen und sieben Einzelsubstanzen mit einer Erkrankung. Die Erkrankungswahrscheinlichkeit stieg um bis zu 90% bei einem Kontakt mit diesen Giftstoffen. Bei den Giftstoffgruppen handelt sich um vier Begasungsgifte und die Gruppe der Chlorkohlenwasserstoffe, bei den Einzelsubstanzen ergab sich ein Zusammenhang bei Aluminiumphosphid, 1,2-Dibromethan (Ethylendibromid), der Phenoxycarbonsäure 2,4,5-Trichlorphenoxyessigsäure, dem Chlorkohlenwasserstoff Dieldrin und der Phosphorsäureester Diazinon, Malathion und Parathion (E 605).
Es gibt aber auch Studien, bei denen keine oder keine signifkanten Zusammenhänge zwischen einer Belastung mit Giftstoffen, in diesem Falle PCB, und Affektstörungen festzustellen waren, jedoch scheinen sie in der Minderzahl zu sein. Die Ergebnisse der Untersuchung einer PCB-belasteten indigenen Mohawk-Gemeinschaft in den USA - der Akwesasne Mohawk Nation - wurde 2012 im Journal of Indigenous Research veröffentlicht. Die indigene Bevölkerung weist dort einen hohen Anteil depressiv Erkrankter auf und die Gebiete um den St.-Lorenz-Strom, in dessen Einzugsgebiet diese Menschen wohnen, gelten aufgrund industrieller Aktivitäten seit den 1940er Jahren bis zum PCB-Verbot als PCB-geschädigt (Quelle: G. S. Morse, G. Duncan et al., Environmental Toxins and Depression in an American Indian Community, Utah State University & University of Washington, 1/2012, Journal of Indigenous Research, Herausgeber Utah State University/Utah & University of North Dakota/Grand Forks 2012, http://digitalcommons.usu.edu/...).
Es wurden die Gesamtmenge an PCB im Blut von 353 Studienteilnehmern bestimmt ohne Unterscheidung einzelner PCB-Verbindungen. Gleichwohl bildeten die Forscher drei PCB-Gruppen zur Verbesserung der Aussagekraft. So wurde zwischen hochchlorierten dioxinähnlichen PCB, hochchlorierten nicht dioxinähnlichen PCB und niederchlorierten PCB differenziert. Die Hypothese lautete, dass ein Zusammenhang zwischen hochchlorierten PCB und der Entstehung einer Depression besteht, die sich jedoch nicht bestätigte. Es konnte lediglich ein nicht signifkanter Zusammenhang zwischen höherchlorierten PCB und einer als höher bewerteten depressiven Symptomatik festgestellt werden.
In der Zusammenfassung der Studienergebnisse wird auf den geringen Grad an Depressivität einer großen Anzahl von Teilnehmern und deren niedriger PCB-Belastung verwiesen, was in der Folge einen Nachweis von Zusammenhängen wesentlich erschwerte. Die Untersuchungsresultate sollten daher mit einer gewissen Skepsis bewertet werden.
Neurologisch-psychiatrische Auswirkungen einer Belastung mit Mikroplastik
(Text folgt)
Fazit: Gift- und Schadstoffe als Verursacher Affektiver Störungen
In Industrie und Landwirtschaft werden zum Teil hochaggressive, neurotoxische Giftstoffe verwendet, die Umwelt ist darüber hinaus auch mit Feinstoffpartikeln belastet. Vor allem eine chronische Belastung mit minimalen Mengen dieser Stoffe kann zu erheblichen Schäden im zentralen und peripheren Nervensystem führen.
Bei vielen Stoffen ist deren Gefährlichkeit noch schwer einzuschätzen und auch das weltweite Verbot der Nutzung von derzeit fast 30 Substanzen befreit nicht von den Auswirkungen der Mengen, die bis zum Verbot dieser stabilen chemischen Verbindungen in die Umwelt gelangt sind, ganz abgesehen von der Tatsache, dass solche Verbote auch umgangen werden können.
Die Gift- und Schadstoffe greifen an unterschiedlichen Stellen funktional oder strukturell Hirnzellen an oder wirken sich nachteilig auf die Hirngefäße aus. Manche wirken wie Mutagene und schädigen die Zell-DNA oder schädigen andere Zellstrukturen, ebenfalls sind negative Auswirkungen auf wichtige Zellprozesse möglich. Die toxischen Mechansimen viele Substanzen sind noch unbekannt oder werden nur teilweise verstanden.
Die Substanzen gelangen entweder über Darm (Nahrung) oder Lunge (Atmung) in den Blutkreislauf und von dort in das Gehirn, einige können über die Atmung das Zentralnervensystem wahrscheinlich auch direkt erreichen, indem sie das Riechzentrum und die Riechnerven als Einfallstor nutzen und sich von dort aus besonders im Gewebe affektrelevanter Hirnareale anreichern. Darüber hinaus bestimmen auch individuelle Faktoren die Gift- und Schadstoffbelastung des Zentralnervensystems, beispielweise der Zustand des Körpers, die familiäre genetische Situation oder der Zustand der entgiftenden Organe Leber und Nieren. Auch periphere Organe, die einen hohen Einfluss auf das psychische Wohlbefinden haben, beispielsweise die Schilddrüse, sind von giftigen Substanzen potentiell bedroht.
Die Gefahr des Entstehens einer Affektiven Störung besteht vor allem dann, wenn Giftstoffe Nerven- und Gliazellen in den affektrelevanten Hirnarealen funktionell oder strukturell schädigen.
In vielen Untersuchungen hat sich bestätigt, dass industriell und landwirtschaftlich genutze Chemikalien bei Menschen auch neurotoxisch wirken können. PCB, das sowohl über den Blutkreislauf als auch über das Riechzentrum das Zentralnervensystem erreicht, führt u. a. zu einem Rückgang der Dopamin-Synthese, Axonschäden oder verlangsamter Reizübertragung. Bei vielen Stoffen wurden negative Auswirkungen auf den Blutfluss im Gehirn festgestellt, Veränderungen der Hirnstromaktivitäten oder sie behindern den Sauerstofftransport im Blut. Feinstaub kann im Körper Entzündungsreaktionen auslösen, ultrafeine Partikel können entzündliche Reaktionen im Gehirn provozieren. Derartige Wirkungen haben das Potential, an der Entstehung einer Affektstörung in der oben beschriebenen Weise mitzuwirken.
In Studien wurden aber auch direkte Zusammenhänge zwischen einer Gift- und Schadstoffbelastung und Affektstörungen festgestellt, von denen hier beispielhaft einige zitiert werden.
In Tierversuchen fand man negative Veränderungen affektrelevanter Hirnstrukturen, in diesem Falle Neuronenveränderungen in den Hippocampi aufgrund einer Fein- und Feinststoffbelastung bei gleichzeitigen Anzeichen für depressives und ängstliches Verhalten.
Darüber hinaus belegen mehrere Untersuchungen, dass eine erhöhte Fein- oder Feinststoffbelastung mit einem erhöhten Suizidrisiko einhergeht.
Arbeitsplatzstudien belegen einen Zusammenhang zwischen der Belastung mit PCB und dem Auftreten verschiedener Affektiver Störungen, Langzeituntersuchungen an Landwirten ergaben Zusammenhänge einer Anwendung von mehreren Pestizide mit der Diagnose einer Depression.
4.13.8 Suchtstoffkonsum und Sucht ▲
Es bestehen keine Zweifel an der Komorbidität von Suchtstoffkonsum/Sucht und psychiatrischen Erkrankungen. Das zeigen Studien, die überdurchschnittliche bis hohe Prävalenzen für das gemeinsame Auftreten beider Merkmale ergaben, insbesondere bei Schizophrenie/Psychosen, Depression/Dysthymie, Bipolarität, Angsterkrankungen und Persönlichkeitsstörungen (Quellen: Suchtforschung & Suchttherapie, Webseite der Universitätsklinik für Psychiatrie und Psychotherapie, Medizinische Universität & Allgemeines Krankenhaus Wien, http://www.sucht‑news.at).
Das bedeutet nicht zwangsläufig, dass zwischen Suchtstoffkonsum/Sucht und psychiatrischen Erkrankungen Kausalzusammenhänge bestehen. Auch wenn vieles dafürspricht, konnten sie bis heute nicht nachgewiesen werden, obwohl sich Medizin und Suchtforschung intensiv darum bemühen.
Das liegt vor allem an fehlenden aussagekräftigen Komorbidiätsmodellen, und solange Forschungen lediglich auf Grundlage drei einfacher und oberflächlicher Basishypothesen (→ folgender Abschnitt) betrieben werden, kann sich daran nichts ändern.
Die komplexe Thematik bedarf einer theoretischen Grundlage, die sämtliche potentiellen Einflussfaktoren berücksichtigt und es ermöglicht, unterschiedliche Szenarien realistisch abzubilden. Mit einigen der in Kapitel 1 und 4 erarbeiteten kausaltheoretischen Hypothesen lässt sich ein aussagekräftiges Gesamtmodell erarbeiten.
Hinweis: Zwar werden die potentiellen Ursachen von Nervenerkrankungen außerhalb von Affektstörungen erst im Kapitel 9 erörtert. Um aber eine Einschätzung von Zusammenhängen zwischen Sucht- und Affekterkrankungen überhaupt machen zu können, muss das im besonderen Fall von Suchterkrankungen teilweise schon an dieser Stelle erfolgen.
Basishypothesen über Suchtstoffkonsum, Sucht und psychiatrische Erkrankungen
In der Suchtforschung dienen drei einfache Basishypothesen der Darstellung möglicher Zusammenhänge zwischen Suchtmittelkonsum/Sucht und psychiatrischen Erkrankungen.
- Basishypothese: Suchtmittelkonsum/Sucht → Psychiatrische Erkrankung
Nach dieser Hypothese ist eine psychiatrische Erkrankung direkte Folge von Suchtstoffkonsum bzw. einer substanzbezogenen Suchterkrankung.
- Basishypothese: Psychiatrische Erkrankung → Suchtmittelkonsum/Sucht
Diese Hypothese ist gegensätzlich zur Basishypothese 1. Erst eine psychiatrische Erkrankung führt zum Gebrauch von Suchtmitteln und zur substanzbezogenen Suchterkrankung.
- Basishypothese: Ursache X → Suchtmittelkonsum/Sucht + psychiatrische Erkrankung
Basishypothese 3 geht von einem korrelativen Zusammenhang zwischen einer psychiatrischen Erkrankung und Suchtstoffkonsum/Suchterkrankung aus, wonach beide Merkmale lediglich auf derselben Ursache bzw. demselben Ursachenbündel beruhen. Ein direkter Kausalzusammenhang besteht demnach nicht.
Eine vierte Aussage verneint jegliche Zusammenhänge und ergänzt die Basishypothesen.
- Koinzidenzannahme: Suchtmittelkonsum/Sucht || Psychiatrische Erkrankung
Suchtmittelkonsum bzw. eine substanzbezogene Suchterkrankung und eine psychiatrische Erkrankung sind völlig unabhängig voneinander (koinzident), es gibt weder gegenseitige Beeinflussungen noch gemeinsame Ursachen.
Die unverbundenen, voneinander unabhängigen Basishypothesen lassen weder eine Darstellung komplexer Zusammenhänge zu, noch benennen sie konkrete Ursachen. Auch differenzieren sie nicht strikt zwischen einem Suchtmittelkonsum und einer Suchterkrankung im eigentlichen Sinne.
Die Hypothesen sind aber gerade wegen ihrer Einfachheit plausibel und als Grundlagen zur Konstruktion eines Komorbiditätsmodells durchaus geeignet, sofern sie kausaltheoretisch interpretiert und miteinander verknüpft werden.
Basishypothesen kausaltheoretisch interpretieren
Potentielle Ursachen der Merkmale Suchtstoffkonsum und Suchterkrankung sind aufgrund ihrer charakterlichen Unterschiede getrennt voneinander zu postulieren.
Für Suchtstoffkonsum, der für sich genommen noch keine Suchterkrankung bedeuten muss, gibt es zwei konkrete Ursachen und eine Koinzidenzannahme:
- Suchtstoffkonsum beruht auf reaktiven Verhaltensanpassungen aufgrund von als unangenehm empfundenen Lebensumständen, beispielsweise psychischem Stress.
- Suchtstoffkonsum beruht auf einer Suchterkrankung.
- Suchtstoffkonsum kann zufälligen Ursprungs sein.
Die primären und sekundären Ursachen von Affekterkrankungen (→ Kapitel 1 und 4) können mit Hilfe eines Analogieschlusses auf Suchterkrankungen übertragen werden. Schon hier wird eine konkrete Verbindung beider Erkrankungen sichtbar:
- Wenn Affektstörungen primär auf neuropathologischen Veränderungen affektrelevanter Hirnstrukturen beruhen (→ Kapitel 1), sollte bei Suchterkrankungen etwas Vergleichbares für suchtregulatorisch relevante Hirnbereiche gelten. Da solche Strukturen nachweislich existieren, ist diese Schlussfolgerung gerechtfertigt. Irrelevant ist, ob sich suchtrelevante Strukturen an bestimmten Stellen konzentrieren oder sich über das gesamte Gehirn erstrecken. Zweitrangig ist, ob Affekt- und Suchtregulierung in denselben Hirnregionen eng miteinander verknüpft sind. Weiter unten wird diese Thematik ausführlicher erörtert (Quelle: dasGehirn.info, Neurowissenschaftliche Gesellschaft e. V., Berlin, https://www.dasGehirn.info).
- Die primär neuropathologischen Veränderungen können sekundär auf Kausalfaktordefiziten (→ Kapitel 4A) und/oder exogenen Noxen (→ Kapitel 4B) beruhen, die in suchtrelevanten Hirnbereichen Schäden verursacht haben, wie sie das in vergleichbarer Weise auch in affektrelevanten Bereichen vermögen. Auch sekundär können damit beide Erkrankungen von denselben Auslösern beeinflusst werden. Interessant ist vor allem die Tatsache, dass Suchtstoffe wegen ihres neurologischen Schadenpotentials ebenfalls zur Gruppe exogener Noxen gehören und in der Lage sind, eine Sucht zu verstärken. Dieser Umstand kann einen Teufelskreis zur Folge haben, bei dem sich Suchtstoffkonsum und Suchterkrankung gegenseitig beeinflussen und verstärken (→ Abbildung 46).
Im Gegensatz zum Suchtstoffkonsum ist bei Suchterkrankungen nicht davon auszugehen, dass diese rein zufällig entstehen. Denn: Jede Erkrankung hat konkrete Ursachen ‑ selbst wenn sie physiologisch bedingt sind. Daher wird auf eine zusätzliche Koinzidenzannahme verzichtet.
Nun werden die Basishypothesen mit Hilfe der obigen Aussagen interpretiert.
- Interpretation von Basishypothese 1 (Suchtmittelkonsum/Sucht → Affekterkrankung):
Der Konsum von Suchtstoffen führt kurz-, mittel- oder langfristig aufgrund negativer Substanzeneigenschaften zu neuropathologischen Veränderungen affektrelevanter Hirnareale und triggert auf diese Weise Entstehung oder Verstärkung Affektiver Störungen. Die Schädigungen erfolgen in Abhängigkeit von Konsumintensität, Konsumdauer und Suchtmittelart auf drei verschiedene Weisen.
- Nach Variante 1.1 schädigen Suchtstoffe die zentralnervöse Affektverarbeitung aufgrund ihrer psychoneuronalen Aktivitäten.
- Bei Variante 1.2 wirken Suchtstoffe neurotoxisch. Beispielsweise bedroht Alkohol das Gehirn durch entzündliche Vorgänge, insbesondere durch Entzündungen von Nerven-, Glia- und Blutgefäßzellen. Geschädigt werden können potentiell sämtliche Hirnregionen, in der hier zu diskutierenden Art und Weise schädigt das Suchtmittel affektrelevante Hirnregionen.
- Suchtsubstanzen können gemäß Variante 1.3 auch periphere Organe, beispielsweise Leber oder Schilddrüsen, negativ beeinflussen, von deren Aktivitäten die Gehirntätigkeit direkt oder indirekt abhängt. Auf diese Weise sind auch für Affekte wichtige Hirnareale gefährdet.
- Interpretation von Basishypothese 2 (Affekterkrankung → Suchtmittelkonsum → Suchterkrankung):
Eine Affektstörung provoziert als Stressfaktor Suchtmittelkonsum durch reaktive Verhaltensanpassungen, mittel- bis langfristig kann das zu einer Suchterkrankung führen. Auch hier gibt es drei Varianten:
- Nach Variante 2.1 führt die Affekterkrankung zu Unsicherheit und Nervosität. Das daraus resultierende Bedürfnis nach Ablenkung und Beruhigung birgt die Gefahr eines Suchtmittelkonsums.
- Nach Variante 2.2 betreibt der Patient ‑ bewusst oder unbewusst ‑ Selbstmedikation, um psychiatrische Symptomatiken mit dem Suchtstoff zu kompensieren, zu verringern oder erträglicher zu machen.
- Nach Variante 2.3 steigt mit anhaltendem Suchtmittelkonsum zusätzlich die Gefahr einer Schädigung suchtregulierender Hirnareale. In Abhängigkeit von Dauer, Intensität und Suchtmittel kann sich eine physische Suchterkrankung entwickeln, ein bisher reaktiver (oder sporadischer) Suchtmittelkonsum wird zur physischen Suchtmittelabhängigkeit. Der Mechanismus von Variante 2.3 ähnelt damit den Varianten 1.1, 1.2 und 1.3, betrifft hier jedoch Veränderungen zentralnervöser Suchtstrukturen. Auch hier sind in analoger Weise drei Untervarianten zu differenzieren. Bei Variante 2.3.1 spielen psychoneuronale, bei Variante 2.3.2 neurotoxische Schädigungen der Suchtstrukturen eine Rolle, bei Variante 2.3.3 sind Schädigungen peripherer Organe die Folgen der Sekundärursache „Suchtmittelkonsum“.
- Interpretation von Basishypothese 3 (Ursache X → Suchtmittelkonsum/Sucht + Affekterkrankung):
Gemäß der Korrelationshypothese können beide Erkrankungen unabhängig voneinander auf derselben Ursache beruhen. Demnach gibt es keine kausalen Zusammenhänge zwischen beiden Erkrankungen. Es ist daher auch unerheblich, zu welchem Zeitpunkt welche Erkrankung auftritt.
Zwei Korrelationsvarianten werden unterschieden: Affekterkrankung und Substanzenkonsum (Variante 3.1) bzw. Affekterkrankung und Suchterkrankung (Variante 3.2).
- Bei Variante 3.1 beruht die Affektstörung ‑ wie immer ‑ auf neurologischen Schädigungen, der beginnende Suchtmittelkonsum basiert unabhängig davon auf einer reaktiven Verhaltensänderung. Als Noxe kommt beispielsweise psychosozialer Disstress infrage. Stress erhöht den Cortisolspiegel, das Stresshormon Cortisol steht im Verdacht, neurologische Schäden zu verursachen. Dies gilt insbesondere bei chronisch erhöhtem Cortisolspiegel aufgrund von langfristigem Stress. Gerade die affektrelevanten Hirnareale reagieren besonders empfindlich auf Cortisol.
- Bei Variante 3.2 ist eine gemeinsame Noxe parallel für die Affektstörung als auch für die langfristige Suchterkrankung verantwortlich. Hier spielt das neurologische Schädigungspotential einer potentiellen Noxe eine Rolle, da beide Merkmale eine neuropathologische Basis haben..
Ein konkretes Beispiel für die Variante 3.2 ist die „Dopamin‑Hypothese“, nach der sowohl Sucht- als auch Affekterkrankung auf einer genetischen Veränderung bzw. Erkrankung beruhen, die Störungen der Dopaminsynthese zur Folge hat.
Bei Variante 3.2 kommt aber auch psychosozialer Stress als gemeinsame Noxe infrage. Im Gegensatz zur Variante 3.1 führt hormoneller Stress parallel zu neurologischen Schädigungen sowohl in affekt- als auch suchtregulatorischen Hirnarealen. Beide Schädigungen müssen nicht unbedingt parallel stattfinden. Auch eine zeitliche Abfolge der Merkmale kann in einer Korrelation begründet sein (→ Abschnitt 3.1).
Völlige Unabhängigkeit der Merkmale Suchtstoffkonsum/Sucht und Affekterkrankung?
Nach der Koinzidenz-Hypothese werden Suchtstoffkonsum/Sucht und eine psychiatrische bzw. affektive Erkrankung völlig unabhängig voneinander ausgelöst.
Beispiel: Die Noxe X ist für den Suchtmittelkonsum (mit‑)verantwortlich ‑ aber nicht für die psychiatrische Erkrankung, während die Noxe Y unabhängig davon ausschließlich die psychiatrische Erkrankung bzw. deren Entstehung triggert. Zwischen den Noxen darf es keinerlei Zusammenhänge geben, ansonsten könnten über Ereignisketten Kausalitäten oder Korrelationen abgeleitet werden.
Auch bei zufälligem Suchtstoffkonsum würde sich nichts ändern, da die Affekterkrankung auf jeden Fall neuropathologisch begründet ist - somit läge auch in diesem Fall eine Koinzidenz vor.
Aufgrund von Studien gibt es Hinweise auf Zusammenhänge zwischen beiden Merkmalen, so dass die Koinzidenz nur in Einzelfällen eine Rolle spielen kann.
Komorbiditätsmodell von Suchtstoffkonsum/Sucht und Affekterkrankungen
Im vierten und letzten Schritt werden alle oben beschriebenen Komorbiditätsphänomene in einem Gesamtmodell integriert; Wechselwirkungen, Kausalketten und sonstige Entwicklungsaspekte werden transparent (→ Abbildung 46).
Das Modell macht die Problematik sichtbar, in dem sich viele Suchtmittelkonsumenten befinden, und die durch folgende Umstände charakterisiert ist:
- Suchtmittelkonsum kann zu neuropathologischen Veränderungen führen und psychiatrische Erkrankungen und/oder Suchtregulationsstörungen zur Folge haben. Psychiatrische Erkrankungen können wiederum das Bedürfnis nach Suchtmittelkonsum befeuern. Suchtmittelkonsum und psychiatrische Erkrankungen triggern sich in einem komplexen Zusammenspiel also gegenseitig, was für Betroffene den Ausbruch aus der Situation schwierig macht. Daher wird die Darstellung als Teufelskreismodell bezeichnet.
- Es ist nicht zwangsläufig eindeutig erkennbar, ob der Suchtmittelkonsum oder die psychiatrische Erkrankung am Beginn der Entwicklung einer Komorbidität steht.
Dabei hätte die (hypothetische) Beantwortung dieser Frage über ihren reinen Informationswert hinaus auch gar keine weitere Bedeutung. Denn der Schluss, beide Erkrankungen wären besser in den Griff zu bekommen, wenn der Behandlungsschwerpunkt auf der Erkrankung läge, die zuerst da war, ist nach dem Teufelskreismodell unzulässig.
- Es gibt zahlreiche innere und äußere Faktoren, die sowohl allgemeine psychiatrische Erkrankungen (einschließlich Affektstörungen) als auch Suchterkrankungen unabhängig voneinander begünstigen.
Da das Modell alle Zusammenhänge nachvollziehbar miteinander verknüpft, ist es bei der Entwicklung individueller Suchtausstiegsstrategien hilfreich. Eine solche Strategie wird im zweiten und dritten Teil diskutiert, in dem kausale Therapiemöglichkeiten gegen affektive Erkrankungen und Sucht erörtert werden (→ Kapitel 6 und 9).
ABBILDUNG 46: SUCHTMITTELKONSUM UND PSYCHIATRISCHE ERKRANKUNG ALS TEUFELSKREIS
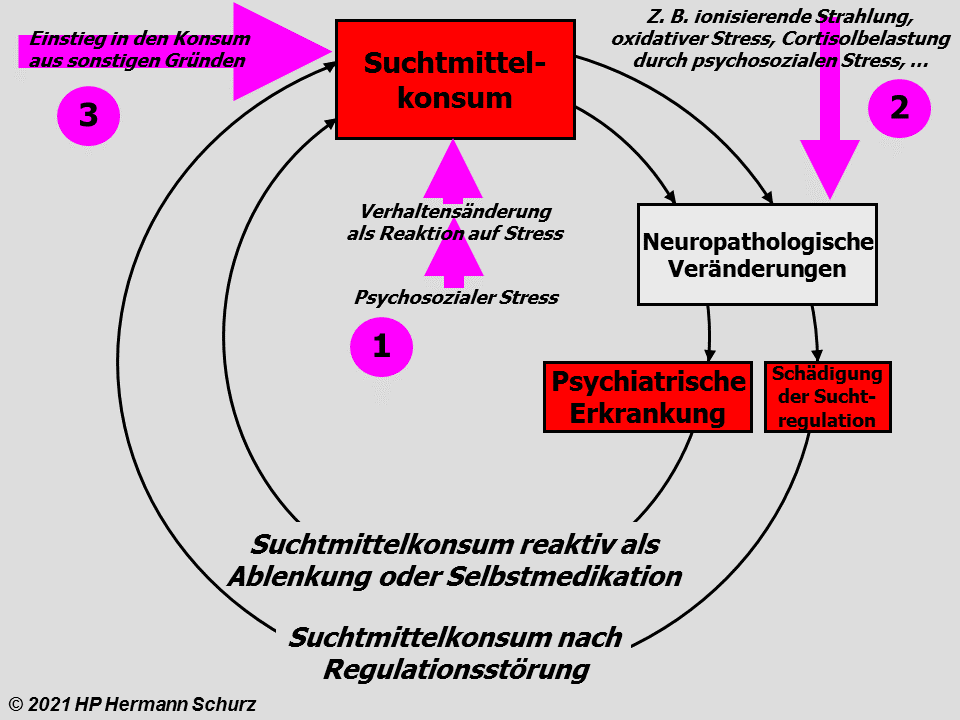
Abbildung 46: Zusammenhänge zwischen Suchtstoffkonsum und einer psychiatrischen Erkrankung sind aufgrund von Wechselwirkungen komplex. Daher kommt zur Darstellung nur die Form der kreisförmigen Kausalkette infrage (→ Abschnitt 3.1). Der Einstieg in den Suchtstoffkonsum hat drei Hauptursachen (Nr. 1 bis 3). Psychosozialer Stress und weitere individuelle Gründe begünstigen den Einstieg in den Suchtmittelkonsum, da sie zu Verhaltensänderungen führen (Nr. 1 und 3). Beim Nr. 2 triggern endogene oder exogene Noxen neuropathologische Veränderungen, die zu einer allgemeinen psychiatrisch-neurologischen Erkrankung führen können, einschließlich affektiver Störungen und einer Suchterkrankung.
Generelle Auswirkungen von Nikotin und Nikotinkonsum
Nikotin wird von der Tabakpflanze als hochwirksames natürliches Insektizid produziert und gehört zu den giftigsten neurotoxischen Substanzen überhaupt. Die für den Menschen tödliche Dosis liegt nach neueren Untersuchungen für Erwachsene bei mindestens 500 mg auf einmal oral aufgenommenem reinen Nikotin. Frühere Angaben über eine letale Dosis von etwa 60 mg haben sich als falsch erwiesen.
Nikotin gelingt fast ausschließlich durch Rauchen von Tabak oder das Inhalieren nikotinhaltiger Liquide in den Körper, während andere Konsumformen (kauen oder schnupfen) eher Ausnahmen sind. Der Rauch einer Zigarette enthält mit ca. ein bis zwei Milligramm wesentlich weniger Nikotin als die ursprüngliche Tabakmenge, die bis zu 13 mg enthält.
Schon wenige Sekunden nach der Aufnahme durch die Alveolen der Lungen erreicht Nikotin über den Blutkreislauf das Zentralnervensystem, denn es durchbricht die Blut-Hirn-Schranke mühelos. Wegen seiner Struktur kann das eigentlich körperfremde Nikotin im Gehirn an bestimmte ‑ meist präsynaptische ‑ Membranrezeptoren binden und diese für körpereigene Botenstoffe und Neuromodulatoren blockieren, für die sie eigentlich vorgesehen sind („Schlüssel-Schloss-Prinzip“). Die Folgen der Blockierung sind veränderte Syntheseraten der betroffenen Botenstoffe in den meist präsynaptischen Nervenzellen.
Membranrezeptoren, an die körperfremdes Nikotin andocken kann, werden als nicotinisch bezeichnet.
Neben neurotoxischen Wirkungen bergen Nikotin und Tabakrauchen weitere Risikofaktoren, die direkt oder indirekt zu einer Schädigung des Zentralnervensystems führen können, insbesondere durch schädliche Auswirkungen auf Blutgefäße. So beeinflusst Nikotin die Gefäßwände negativ und führt kurzfristig zu einer Gefäßverengung und langfristig zu chronischen Gefäßschäden, bei denen auch die Elastizität von Arterien und Venen abnimmt. Durch Nikotin und die vielen Schadstoffe des Tabaks erhöht sich oxidativer Stress in den Blutgefäßzellen, und es kann zu weiteren chronischen Schäden an Gefäßwänden kommen. Davon sind alle Gefäßarten betroffen, neben Arterien und Venen auch Arteriolen oder kleinste Kapillargefäße. In der Folge werden Durchblutung und Mikrozirkulation behindert, so dass die Nerven- und Gliazellen nicht mehr adäquat mit Nährstoffen und Sauerstoff versorgt werden und auch die Gefahr eines Schlaganfalls steigt. Ein weiterer Aspekt ist die Schädigung der Lungenfunktionen durch Rauchen und damit einhergehende Sauerstofffehlversorgungen. Gerade eine ausreichende Sauerstoffversorgung ist für die Funktionsfähigkeit des Gehirns entscheidend.
Solche speziellen Gesichtspunkte des Rauchens werden in anderen Abschnitten thematisiert. Die Mechanismen und Folgen oxidativen Stressen sind zum Beispiel Inhalte des Abschnitts 4.13.6, die Auswirkungen von Gefäßschäden werden im Hauptabschnitt 4.14 erörtert (→ Vaskuläre Erkrankungen).
Nikotinwirkungen im Detail
Für die Steigerung von Wachheit und geistiger Leistungfähigkeit durch Nikotoin ist vor allem der nicotinische Acetylcholin‑Rezeptortyp von Bedeutung, der eigentlich für die Bindung des Neurotransmitters Acetylcholin (ACh) vorgesehen ist und vom körperfremden Nikotin blockiert wird.
ACh-Rezeptoren befinden sich in großer Zahl vor allem präsynaptisch im cholinergen System, das ein zusammenhängendes System von Nervenzellen im Gehirn bezeichnet, die alle ‑ unter anderem ‑ Acetylcholin als Botenstoff verwenden. Damit sind auch affektrelevante Areale betroffen. Nikotinaffine ACh-Rezeptoren findet man auch im restlichen Körper in der gestreiften Muskulatur, in den autonomen Ganglien des peripheren Nervensystems und im Nebennierenmark.
Darüber hinaus kommen nicotinische Rezeptoren auch in anderen Neurotransmittersystemen des Zentralnervensystems vor, beispielsweise im GABA-nergen, dopaminergen, glutamatergen oder serotonergen System.
Nikotin beeinflusst durch diese vielfältigen Auswirkungen die Freisetzung von Acetylcholin, beta-Endorphin, Dopamin, GABA, Glutamat, Noradrenalin, Serotonin und wahrscheinlich noch weiteren neuroaktiven Substanzen. In den meisten Fällen sind affektrelevante Hirnareale mitbetroffen.
Auf dieser physiologischen Wirkungsvielfalt sollen die mannigfaltigen Wirkungen des Nikotins auf das Befinden und die Psyche eines Konsumenten resultieren. (Quelle: Nikotin - Pharmakologische Wirkung und Entstehung der Abhängigkeit ‑ Fakten zum Rauchen, Deutsches Krebsforschungszentrum Heidelberg, Stabsstelle Krebsprävention und WHO-Kollaborationszentrum für Tabakkontrolle, Heidelberg 2008, https://www.dkfz.de/...).
Es soll im Zusammenhang mit der Blockierung der jeweiligen Rezeptoren und der erhöhten Freisetzung des dazugehörigen Botenstoffs zu folgenden Nikotinwirkungen beim Menschen kommen:
- Acetylcholin - Wachheit und Steigerung geistiger Leistungsfähigkeit
- beta-Endorphin - Minderung von Angst und Stress
- Dopamin - Wohlgefühl, Belohnung und Appetitunterdrückung
- GABA - Minderung von Angst und Spannung
- Glutamat - Lernen und Gedächtnisverbesserung
- Noradrenalin - Wachheit und Appetitunterdrückung
- Serotonin - Stimmungsregulation und Appetitunterdrückung
Durch eine Spirale typischer Suchtmechanismen kann Nikotingenuss bei Konsumenten sowohl in einer psychischen als auch einer körperlich-physischen Abhängigkeit münden.
Bei der Entstehung der körperlich-physischen Nikotinabhängigkeit soll der Botenstoff Dopamin die herausragende Rolle spielen. Die Abhängigkeit entsteht wahrscheinlich in zwei Schritten. Zunächst stimuliert Nikotin zusätzliche Neurotransmitterausschüttungen. Der erhöhte Neurotransmitterspiegel weckt dann ein weiteres Verlangen nach Nikotin. Immer größere Nikotinmengen bewirken langfristig eine Veränderung der Anzahl nicotinischer Rezeptoren und der Körper verlangt nun ständig und auch nach immer höheren Mengen Nikotins.
Geht ein Konsument diesem Verlangen nicht nach, stellen sich Entzugserscheinungen ein: Unruhe, Gereiztheit, Ängste, Konzentrationsschwierigkeiten und eine depressive Verstimmung, ggf. kann sich eine schon bestehende Depression verstärken.
Relativ leicht nachvollziehen lässt sich dieser als Toleranzbildung bezeichnete Effekt am Beispiel der durch Nikotin beinflussbaren Dopamin-, GABA- und Glutamatfreisetzung (→ Abbildung 47).
ABBILDUNG 47: TOLERANZBILDUNG AM BEISPIEL DREIER BOTENSTOFFE
Abbildung 47: Der Neurotransmitter Dopamin steht im Mittelpunkt der Toleranzbildung und Suchtentstehung, dessen Synthese durch Nikotingenuss besonders stark stimuliert wird. Nikotin bindet im ventral-tegmentalen Areal des Mittelhirns (VTA) an die nACh-Rezeptoren dopaminbildender Nervenzellen in der Substantia nigra, die daraufhin vermehrt Dopamin freisetzen. Der zum dopaminergen System gehörende Nucleus accumbens (NA) der Basalganglien, das sogenannte Belohnungszentrum, wird durch die Dopaminbahnen des VTA stärker gereizt (rote Pfeillinien vom VTA zum NA) und erhöht ebenfalls die Dopaminausschüttung. Darüber hinaus setzt der NA nikotinbedingt verstärkt GABA frei, das wiederum auf das VTA rückwirkt (blaue Pfeillinien vom NA zum VTA) und die Dopaminausschüttung im VTA nochmals steigert. Die nun wesentlich erhöhte Dopaminausschüttung im VTA des Mittelhirns wirkt sich auch auf den präfrontalen Cortex aus (rote Pfeillinie vom VTA zum Cortex). Eine nikotinbedingte Reaktion des präfrontalen Cortex besteht darüber hinaus in der Freisetzung von Glutamat, das den NA des Belohnungszentrums zusätzlich stimuliert und die Dopamin-Synthese nochmals erhöht (grüne Pfeillinien zum NA). Alle Vorgänge betreffen in erster Linie affektrelevante Hirnareale.
Mindestens zwei weitere Tabaksubstanzen führen zu einer Steigerung der Monoamin-Synthese
Das Inhalieren von Tabakrauch erhöht die Anzahl einiger Neurotransmitter mindestens auch auf folgende Weisen:
- Das dem Tabak zugesetzte Ammonium erhöht die Nikotinwirkungen, indem es die Dopamin-Synthese ankurbelt.
- Beim Verbrennen des im Tabak enthaltenen Zuckers entsteht Acetaldehyd. Das behindert die Produktion von Monoaminoxidase-Hemmern in den Nervenzellen und erhöht nochmals die Anzahl der Monoamine in den synaptischen Spalten, beispielsweise auch von Dopamin.
Dabei enthält Tabak mehr als 4.000 chemische Substanzen, und nur über einen Bruchteil davon gibt es Kenntnisse deren zentralnervöser Wirkungen. Es ist davon auszugehen, dass sich ein Teil dieser Substanzen ebenfalls auf den Gehirnstoffwechsel auswirkt und auch mit Nikotin interagiert.
Zusammenhänge zwischen Nikotinkonsum, Nikotinsucht und Affektstörungen
Nikotinwirkungen betreffen vor allem affektrelevante Hirnareale, so dass Zusammenhänge zwischen Nikotinkonsum und Affektstörungen plausibel sind.
Diese Zusammenhänge sind auch empirisch nachgewiesen. Einen Überblick über das Thema gibt der Psychiater Georg Winterer in einem Aufsatz aus dem Jahre 2013 (Quelle: Georg Winterer, Rauchen und psychiatrische Erkrankungen: Ein Überblick, Journal für Neurologie, Neurochirurgie und Psychiatrie, 14 (3), S. 119, Krause & Pachernegg GmbH, Verlag für Medizin und Wirtschaft, Gablitz/Österreich, 2013, http://www.kup.at/...). Bei den diesem Artikel zugrundeliegenden Untersuchungen wurde Rauchen generell betrachtet, also nicht nur ein isolierter Nikotinkonsum.
Die Ergebnisse zahlreicher Studien ‑ der Beitrag umfasst 91 Quellenangaben ‑ deuten auf Zusammenhänge zwischen den Merkmalen, nach Aussage von Winterer konnte jedoch kein eindeutiger Beziehungscharakter nachgewiesen werden, zum Beispiel mit einem unbekannten Faktor („hidden variable“). Ein ausführlicher Überblick über die empirische Forschung folgt noch gegen Ende des Abschnitts, hier vorab einige Erkenntnisse aus Winterers Beitrag:
- 1/4 bis 1/3 aller Nikotinabhängigen haben diagnostizierte psychiatrische Störungen.
- In den USA konsumiert der 7%ige Bevölkerungsanteil mit psychiatrischen Störungen etwa 34% aller Zigaretten.
- In westlichen Ländern sind Psychiatriepatienten überdurchschnittlich starke Raucher. 60% der depressiven Patienten sind nikotinabhängig, bei Schizophrenie steigt dieser Anteil auf bis zu 90%. Innerhalb der Gesamtbevölkerung beträgt der Anteil dagegen nur 30%.
- Auf die gesamte Lebenszeit betrachtet haben Raucher ein dreifach höheres Risiko, an einer Depression zu erkranken (Lebenszeitprävalenz).
- Während der ersten drei Monate eines Rauchentzugs besteht ein erhöhtes Risiko, an einer depressiven Störung zu erkranken, vor allem, wenn das Rauchen im jugendlichen Alter begonnen wurde.
- Raucher haben ein höheres Suizidrisiko, auch unabhängig von einer depressiven Erkrankung oder sonstigem Substanzenmissbrauch. Bei Nikotinabhängigen ist dieses Risiko achtfach erhöht.
- Auch andere psychiatrische Störungen, beispielsweise Angststörungen, Aufmerksamkeitsdefizitsyndrome oder posttraumatische Belastungsstörungen, und Abhängigkeitserkrankungen, beispielsweise Alkoholabhängigkeit, gehen mit erhöhten Prävalenzraten von Nikotin- bzw. Tabakabhängigkeit einher.
- Der Anteil stark rauchender psychiatrisch vorbelasteter Patienten steigt im Vergleich zur Gesamtbevölkerung westlicher Industrienationen stetig. Betrug der Anteil von Rauchern im Jahre 1960 noch 43%, konnte er durch verschiedene Informations- und Gesundheitskampagnen auf 23 bis 30% gesenkt werden. Bei dem Anteil, den Psychiatriepatienten ausmachen, waren in dieser Richtung keine Erfolge zu verzeichnen.
Konkrete Ursachen der Zusammenhänge zwischen Nikotinkonsum und Affektstörungen lassen sich ableiten, indem die Basishypothesen A, B und C interpretiert werden, zum Beispiel mit Hilfe der kausaltheoretischen Modelle (→ Kapitel 1). Eine solche Vorgehensweise wurde schon weiter oben suchtmittelunabhängig durchgeführt (→ Abschnitt über die realen Ursachen der Zusammenhänge zwischen Suchtmittelkonsum und Affektstörungen). Es ergeben sich beim Sonderfall des Nikotins fünf Szenarien:
- Nikotinkonsum verursacht Affektstörungen
Primär ist der Nikotinkonsum, sekundär die Affektstörung. Nikotin ist an der Genese der Erkrankung aktiv beteiligt. Diese Vermutung folgt damit Basishypothese A.
Da eine Affektstörung aus kausaltheoretischer Sicht immer einen neurobiologischen (= neurologischen) Hintergrund hat, müsste Nikotin demnach in der Lage sein, durch neurobiologische Veränderungsprozesse eine Affektstörung mindestens mitzuverantworten.
So könnte eine langfristige Desensitivierung nikotinischer Acethylcholin-Rezeptoren die Acethylcholin-Übertragung behindern und auf diesem Wege die Entstehung einer Depression (oder die Verstärkung einer bestehenden Erkrankung) begünstigen.
Ebenfalls ist eine nikotinbedingte Erschöpfung der Dopamin-Synthese denkbar, wodurch eine Affektstörung begünstigt würde. Auch negative Beeinflussungen weiterer Botenstoffe durch Nikotin, die bei der Affektverarbeitung eine Rolle spielen, kämen als Ursachen infrage.
- Affektstörungen provozieren den Nikotinkonsum
Primär ist hier die Affektstörung, zum Beispiel eine Depression oder Bipolare Störung, die den Tabakkonsum triggert (Basishypothese B).
Erkrankungsbedingte Nervosität oder das Bedürfnis nach Ablenkung könnten für den Tabakkonsum (mit‑)verantwortlich sein.
Diese rein verhaltensbedingte Erklärung ist unabhängig von neurobiologischen Veränderungsprozessen plausibel. Aber auch hier ist zumindest ein neurobiologischer Hintergrund für die Verhaltensänderung denkbar. So könnte der affektiv Erkrankte der Versuchung nachgeben, seine auf neurologischen Krankheitsprozessen basierende affektive Symptomatik mit Hilfe des Suchtstoffs Nikotin durch Selbstmedikation zu kompensieren.
Geht man beispielsweise von einem dysfunktionalen dopaminergen Belohnungssystem mit einer zu geringen Dopamin-Synthese als eine der Ursachen der Affektstörung aus, könnte sich die Dopamin-Syntheserate durch Nikotin ‑ zumindest für eine gewisse Zeit ‑ wieder erhöhen, so dass die affektiven Symptome als erträglicher empfunden werden.
Ähnliche suchtfördernde Prozesse sind auch bei den anderen Botenstoffen Acethylcholin, beta-Endorphin, GABA, Glutamat, Noradrenalin und/oder Serotonin vorstellbar, die auch parallel stattfinden können.
- Affektstörungen führen zur Chronifizierung des Nikotinkonsums (Nikotinsucht)
Ein (moderater) Nikotinkonsum bestand schon vor der Affekterkrankung. Die Erkrankung führt jedoch dazu, dass Bedürfnis und Fähigkeit, das Rauchen zu beenden, stark gemindert sind und Nikotinsucht droht. Erklärungsvariante 3 ähnelt damit Erklärungsvariante 2 und folgt ebenfalls Basishypothese B.
Reaktiv-verhaltensbedingt könnten Nervosität und das Bedürfnis nach Ablenkung, die beide auf die Erkrankung zurückzuführen sind, für die Chronifizierung des Nikotinkonsums (mit‑)verantwortlich sein, auch hier unabhängig von neurologischen Erkrankungs- bzw. Veränderungsprozessen.
Ebenfalls ist ein neurobiologischer Hintergrund denkbar. Nikotin diente dann der Selbstmedikation, um die neurobiologischen Auswirkungen der Affektstörung zu kompensieren und abzumildern. Daher ist das Bedürfnis des Tabakrauchens nur schwer bzw. gar nicht zu beeinflussen und zu drosseln. Die Chronifizierung des Rauchens scheint unausweichlich.
Die Varianten 2 und 3 treten naturgemäß häufig hintereinander auf.
- Nikotinkonsum und Affektstörungen korrelieren miteinander
Nikotinkonsum und Affektstörung beruhen nach Basishypothese C auf einer oder mehreren gemeinsamen Ursachen.
Reaktiv-verhaltensbedingt könnte eine beliebige Stresssituation als Ursache den Nikotinkonsum verantworten. Wie bei Variante 2 bzw. 3 kommen dafür Nervosität und/oder das Bedürfnis nach Ablenkung von der Stresssituation infrage. Die Affektstörung hat dagegen immer einen neurologischen Hintergrund.
Variante 4 kann auf mehreren sehr unterschiedlichen neurobiologischen Prozessen beruhen.
So ist beispielsweise die Annahme einer polygenetischen Veranlagung für die beiden Merkmale Niktonsucht(‑anfälligkeit) und Anälligkeit für eine Affekterkrankung diskussionswürdig. Die Möglichkeit einer Korrelation träfe allerdings nur zu, wenn exakt dieselbe polygenetische Abweichung sowohl für die Tabaksucht als auch die Affektstörung verantwortlich ist, was natürlich sehr unwahrscheinlich und darüber hinaus nicht zu belegen ist. Sind aber unterschiedliche polygenetische Einflüsse für das jeweilige Merkmal verantwortlich, läge streng genommen keine Korrelation vor.
Darüber hinaus ist zu hinterfragen, auf welche Art und Weise die polygenetischen Abweichungen überhaupt zu beiden Merkmalen führen (→ Abschnitt 4.7). Beispielsweise könnte die spezielle Genkombination die Entstehung einer bestimmten Hirnstruktur beeinflussen, die dann sowohl eine Affektstörung als auch Tabaksucht fördert. Damit läge aber eigentlich in der speziellen Hirnstruktur die direkte gemeinsame Ursache, die polygenetische Veränderung könnte man demgegenüber als Primärursache bezeichnen. Hier ist eine Kausalkette mit mehreren aufeinander folgenden Ursachen und Wirkungen Teil der Erklärung.
Aber auch die schon erörterte Erklärungsvariante 2 verliert unter dem Gesichtspunkt einer möglichen Korrelation ihre Eindeutigkeit. So könnte der Tabakkonsum zeitlich zwar auf eine affektive Erkrankung folgen, beides könnte aber letztlich auf denselben hirnorganischen Auffälligkeiten beruhen. Die zeitliche Differenz zwischen beiden Merkmalen wäre rein zufällig. Rauchen als Selbstmedikation und Affektstörungen ständen aus dieser Perspektive aus gesehen in einem korrelativen ‑ und nicht ursächlichen ‑ Verhältnis zueinander. Es könnte aber auch umgekehrt sein, dass der Betroffene zunächst mit dem Rauchen beginnt und anschließend eine Affekterkrankung erleidet. Hier könnte dann der gegensätzliche Eindruck entstehen, es träfe Erklärungsvariante 1 zu und das Rauchen sei ursächlich für die Erkrankung (mit-)verantwortlich. Dennoch läge auch in diesem Falle eine Korrelation vor, denn auch in diesem Beispiel ist der zeitliche Abstand zufällig und suggeriert lediglich einen Kausalzusammenhang.
- Tabakkonsum und Affektstörungen stehen miteinander in Wechselwirkungen
Vier verschiedene plausible Erklärungsvarianten mit einer teilweise unklaren Gemengelage mehrerer Ursache-Wirkungs-Szenarien deuten sehr stark auf komplexe Wechselwirkungen zwischen Tabakkonsum und einer Affektstörung hin, die hier im Erklärungsmodell 5 miteinander vereint werden. Diese Ansicht spiegelt sich im Teufelskreismodell wider, das zu Beginn des Abschnitts über Suchtstoffe diskutiert wurde.
Auch die Sichtweise der Affektstörung als einer sich aus einem multifaktoriellen Geschehen heraus entwickelnden Erkrankung spricht für dieses Modell und gegen einseitige bilaterale Zusammenhänge.
Als weiteres Beispiel soll hier die wissenschaftliche Diskussion dienen, eine fehlregulierte Stressverarbeitung könne einen hohen Nikotinkonsum (mit‑)verantworten. In diesem Falle versuche der Patient durch Selbstmedikation seine erhöhte Stresssensitivität mit Hilfe des Nikotins zu regulieren.
Aber gerade hier sind Zusammenhänge zwischen Stressproblematik, Nikotinsucht und einer Affektstörung auf unterschiedliche Weisen vorstellbar, beispielsweise wenn die in Abschnitt 4.12.1 erläuterten Stressmodelle zugrundegelegt werden (→ Kapitel 4 A). Stressbedingte ungünstige hirnorganische Veränderungen könnten danach sowohl die Affekterkrankung also auch die Sucht parallel auslösen, so dass eine Korrelation zwischen beiden Merkmalen vorläge. Aber auch ein mehrstufiger kausaler Zusammenhang (Kausalkette) kommt infrage, nämlich wenn Stress zu hirnorganischen Veränderungen führt, die wiederum den Ausbruch einer Affektstörung verursachen bzw. mitverursachen.
Ergebnis: Zwischen Tabakkonsum und Affekterkrankungen besteht mit größter Wahrscheinlichkeit ein komplexer multilateraler Zusammenhang, der mit dem Teufelskreismodell realistisch beschrieben ist (→ Abbildung 48 unten bzw. Abbildung 46 oben). Der Begriff „Teufelskreis“ beschreibt genau das Dilemma, in dem sich Nikotinabhängige befinden. Dabei ist die Frage, was zuerst da war ‑ Nikotinsucht oder Affekterkrankung ‑ letztlich unerheblich. Eine solche Frage führt zu unproduktiven Auseinandersetzungen um eine „Schuldfrage“, die Betroffenen nicht weiterhelfen.
Lediglich die Problematik von durch einen Nikotinentzug ausgelöste Affektstörungen lässt sich in das Teufelskreismodell nicht integrieren und muss daher parallel beachtet werden.
Vor allem unter kausaltheoretischen Gesichtspunkten ist das Teufelskreismodell die einzige plausible Erklärung der komplexen Zusammenhänge zwischen Tabakrauchen und Affektstörungen. Das Modell schließt nicht aus, dass ein Einstieg in die Nikotinsucht auch zufällig bzw. völlig anderen Gründen geschehen kann.
Neben der Kausalitätsfrage hat das Thema noch weitere Gesichtspunkte. So ist die Frage bisher nicht klar beantwortet, ob es sich bei den Schäden durch Nikotin um kurz- oder langfristige Schäden handelt und wie sich eine unterschiedliche Schadendauer auf die Anfälligkeit für Affektstörungen auswirkt.
Derzeit wird angenommen, dass sich die Veränderungen an den nicotinischen Acethylcholin-Rezeptoren nach erfolgreicher Tabakentwöhnung in allen Gehirnarealen wieder vollständig zurückbilden, also nicht von langfristigen Schäden durch Nikotin auszugehen ist. Die Frage ist jedoch, ob das in jedem individuellen Fall zutrifft bzw. ob und welche Ausnahmen es gibt. So stellen die Acethylcholin-Rezeptoren beispielsweise nur einen ‑ wenn auch wichtigen ‑ Rezeptortypen dar, der durch Nikotin stimuliert und in seiner Anzahl verändert wird. Das darf jedoch nicht auf andere neurologische Mechanismen übertragen werden.
Darüber hinaus ist auch die Frage interessant, wie sich die Nikotinwirkungen im jugendlichen Gehirn von denen Erwachsener unterscheiden, denn die Entwicklung des Zentralnervensystems ist erst etwa in der Mitte des dritten Lebensjahrzehnts abgeschlossen (→ Abbildung 40 über die Entwicklung des Gehirns in Kinheit und Jugend). Sind rauchende Jugendliche dadurch stärker gefährdet?
Auch potentielle Folgen des Rauchens während der Schwangerschaft sind eine weiteres Themengebiet. Es ist erwiesen, dass sich Rauchen unter anderem ungünstig auf die Gehirnentwicklung eines Fötus auswirkt, mit dem später unter anderem auch eine gesteigerte Anfälligkeit für Affektstörungen einhergehen könnte.
ABBILDUNG 48: TABAKSUCHT UND AFFEKTERKRANKUNG ALS TEUFELSKREIS
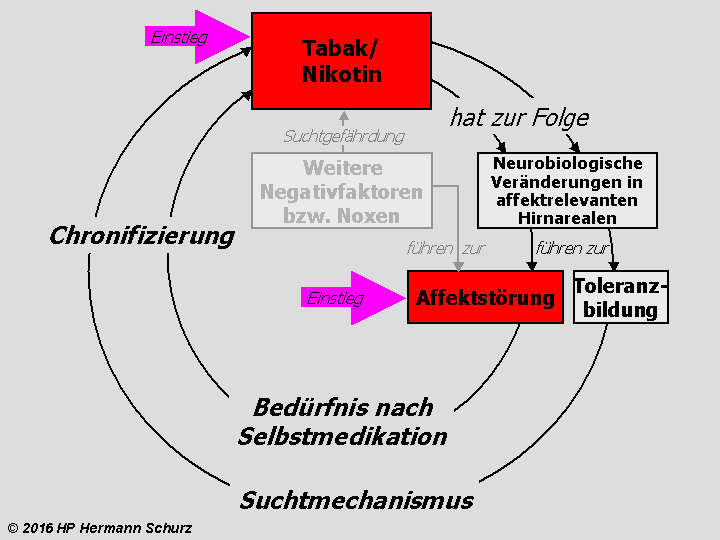
Abbildung 48: Zusammenhänge zwischen Tabakrauchen bzw. Nikotin und einer Affektstörung sind komplex und lassen sich nicht auf eindimensionale Kausalitäten reduzieren. Es besteht eine komplizierte Wechselwirkung zwischen Nikotin und Affektstörung, die Betroffene in einen Teufelskreislauf führen kann. Mit dem Modell einer kreisförmigen Kausalkette lässt sich diese Komplexität vereinfacht darstellen. Innerhalb der kreisförmigen Kausalkette ist die Bestimmung einer absoluten Kausalität nicht möglich (→ Abschnitt 3.1 über absolute oder relative Kausalitäten und kreisförmige Kausalketten). Der Einstieg in die Nikotinsucht kann zufällig oder durch unterschiedliche Negativfaktoren, zum Beispiel aufgrund der leichten Verfügbarkeit und gesellschaftlichen Toleranz des Tabakkonsums, aber auch einer individuellen Suchtgefährdung geschuldet sein, die beispielsweise auf polygenetischen Faktoren beruht, die wiederum mit hirnorganischen Auffälligkeiten einhergehen. Eine Affekterkrankung kann durch Nikotin mitausgelöst oder eine schon bestehende Erkrankung verstärkt werden. Über das Bedürfnis nach Selbstmedikation mit Hilfe des Nikotins ist ein alternativer Einstieg in die Sucht möglich. Mittel- bis langfristig kann Nikotin die Erkrankung durch neurobiologische Prozesse nicht nur verstärken, sondern parallel auch zur Toleranzbildung beitragen und damit die Chronifizierung der Sucht auf doppelte Weise fördern. Einmal im Kreislauf gefangen, ist die Situation für Betroffene unabhängig von der Einstiegsursache für den Tabakkonsum vergleichbar.
Nach diesen eher theoretischen Ausführungen ist nun zu analysieren, welche Antworten die Empirie auf die Kausalitätsfrage bei Tabakkonsum bzw. Nikotinsucht und Affektstörungen geben kann. Mit den schon weiter oben im Beitrag von Georg Winterer aufgeführten Studien wurden zwar Zusammenhänge nachgewiesen, jedoch lassen sie nach Aussage des Autors keine Schlussfolgerungen über deren Charakteristik zu. Mit ihnen ist daher nicht zu belegen, ob es sich um kausale oder korrelative Beziehungen handelt (Quelle: Georg Winterer, Rauchen und psychiatrische Erkrankungen: Ein Überblick, Journal für Neurologie, Neurochirurgie und Psychiatrie, 14 (3), S. 119, Krause & Pachernegg GmbH, Verlag für Medizin und Wirtschaft, Gablitz/Österreich, 2013, http://www.kup.at/...).
Leider sind Untersuchungen, die sich mit der Kausalitätsfrage beschäftigen, relativ rar. Anfang 1993 erschienen in der Fachzeitschrift Archives of General Psychiatry die Ergebnisse einer im Jahre 1992 von Kenneth Kendler und einem Team durchgeführten umfangreichen Zwillings-Studie, mit der nach Belegen für einen ursächlichen Zusammenhang zwischen Tabakkonsum und der Enstehung einer Depression gesucht wurde (Quelle: Kenneth S. Kendler, Michael C. Neale, Ronald C. Kessler et al., Smoking and Major Depression ‑ A Causal Analysis, Archives of General Psychiatry/JAMA Psychiatry, Vol. 50, 1/1993, American Medical Association, Chicago/Illinois, USA, https://www.ncbi.nlm.nih.gov/pubmed/...).
Die Grundlage der Untersuchung waren 1.566 speziell ausgewählte eineiig-monozygotische und zweieiig-dizygotische weibliche Zwillinge, deren täglicher Tabakkonsum nachgewiesen in einem starken Zusammenhang mit dem Auftreten einer Major Depression stand. Mit der statistischen Cotwin-Untersuchungsmethode sollte nun herausgefunden werden, ob dieser Zusammenhang kausal oder nicht-kausal ist.
Die Untersuchungen ergaben bei den Studienteilnehmerinnen keinen kausalen Zusammenhang zwischen Rauchen und einer Major Depression. Die Autoren kamen jedoch zu dem Schluss, dass familiäre Faktoren ‑ in diesem Falle polygenetische Faktoren ‑ beide Merkmale prädisponieren.
Damit läge eine Korrelation vor, wobei die schon weiter oben erörterte Frage jedoch unbeantwortet bleiben muss, ob dieselben polygenetischen Faktoren für beide Merkmale verantwortlich sind oder unterschiedliche polygenetische Faktoren, denn nur im ersten Falle dürfte man von einer echten Korrelation sprechen.
Die häufig vertretene Selbstmedikations-Hypothese, die durch zahlreiche Aussagen von Nikotinkonsumenten gestützt wird, Tabakrauchen hätte positive Effekte auf ihr affektives Erleben, wurde im Jahre 2007 in einer von der US-amerikanischen Psychiaterin Bonnie J. Spring geleiteten und am Edward Hines Jr. Veterans Administration Hospital in Hines/Illinois (USA) durchgeführten Doppelblind-Studie geprüft und teilweise bestätigt (Quelle: Bonnie Spring, Bradley Appelhans, Donald Hedeker et al., Nicotine effects on affective response in depression-prone smokers, Psychopharmacology, Vol. 196 (3), 2008, S. 461 ‑ 471, https://www.ncbi.nlm.nih.gov/...). Die 165 relativ jungen Raucher im dritten und vierten Lebensjahrzehnt wurden in drei Gruppen eingeteilt. Davon hatten 63 Personen im Lebensverlauf niemals eine Major Depression (MDD) als Diagnose, 61 Personen konnten eine MDD-Diagnose in ihrer Vergangenheit vorweisen, waren zum Zeitpunkt der Studie jedoch symptomfrei, und bei 41 Personen wurde sowohl aktuell als auch in der Vergangenheit eine MDD diagnostiziert.
Die Teilnehmer wurden in einen negativen bzw. positiven Stimmungszustand versetzt und rauchten anschließend nikotinhaltige oder nikotinfreie Zigaretten, ohne dabei über den Niktongehalt aufgeklärt zu werden und auch das Studienpersonal wusste darüber nicht Bescheid (Doppelblind-Prinzip).
Die Forscher wollten feststellen, welchen Einfluss nikotinhaltige Zigaretten auf einen zuvor stimulieren Gemütszustand haben: Sind sie in der Lage, eine positive Stimmung zu verstärken bzw. eine negative Stimmung aufzulösen? Hängt die Stärke solcher Effekte von der Anfälligkeit der jeweiligen Person für eine Depression ab und sind solche Effekte dann besonders stark, wenn die Person zum Zeitpunkt der Studie als depressiv diagnostiziert ist?
Die Ergebnisse wiesen darauf hin, Nikotin könne bei depressionsanfälligen Menschen als stimmungsverstärkender Faktor einer bewusst ausgelösten positiven Grundstimmung fungieren. Eine positive Stimmungslage konnte besonders bei den als depressiv diagnostizierten Personen durch eine nikotinhaltige Zigarette verstärkt werden, ebenfalls lösten sich bei ihnen bestehende negative Stimmungen auf. Die Gruppe mit einer früheren MDD-Diagnose konnten eine positive Grundstimmung mit einer nikotinhaltigen Zigarette ebenfalls verstärken, obwohl der Effekt im Vergleich mit der anderen Gruppe nicht so intensiv war.
Aufgrund dieses Ergebnisses war zu erwarten, dass man mit Nikotin bei einem zuvor negativ stimulierten Gemütszustand zu vergleichbaren Resultaten kommt und in der Lage ist, die negativen Gefühle zu zerstreuen. Interessanterweise war das Gegenteil der Fall - und dies bei allen drei Gruppen: Die Stimmung verschlechterte sich unter dem Einfluss des Nikotins.
Damit ergibt sich aufgrund der Ergebnisse dieser Studie für die Selbstmedikations-Hypothese keine einheitliche Argumentationsgrundlage. Es scheint zwar so zu sein, dass Nikotin bei depressionsanfälligen Menschen positive Gefühle verstärken kann, jedoch eine schlechte Grundstimmung bei allen Personen weder aufzulösen noch erträglicher zu machen vermag, sondern den negativen Gemütszustand noch verschlimmert.
Es sind jedoch die Studienlimitationen zu beachten. Es wurden keine weiteren Nikotinwirkungen untersucht. Die Arbeit von Spring et al. bezieht sich ausschließlich auf den Stimmungszustand. Positive Auswirkungen auf die Konzentrationsfähigkeit, Wachheit oder angstlösende Eigenschaften wurden nicht berücksichtigt.
Eine mit der Spring-Studie vergleichbare Untersuchung mit 104 nikotinabhängigen Rauchern, die im Jahre 2010 von Kenneth A. Perkins und einem Team an der University of Pittsburg/USA durchgeführt wurde, kam hinsichtlich der Fähigkeit des Nikotins, schlechte Stimmungen aufzulösen, zu einem ähnlichen Ergebnis (Quelle: Kenneth A. Perkins, Joshua L. Karelitz, Grace E. Giedgowd et al., Acute Negative Affect Relief from Smoking Depends on the Affect Situation and Measure but Not on Nicotine, Departments of Psychiatry and Psychology, University of Pittsburgh, Pittsburg/Pennsylvania, USA, Biological Psychiatry, 2010 (67), S. 707 ‑ 714, Elsevier B. V., Amsterdam/NL 2010), http://www.biologicalpsychiatryjournal.com/...).
Perkins und sein Team fanden heraus, dass Rauchen nur unter der Bedingung einer eintägigen Abstinenz negative Stimmungen unterschiedlicher Ursachen auflösen kann. Entzugserscheinungen scheinen daher eine große Rolle zu spielen. Unterblieb die Abstinenz, hing der Rückgang der negativen Affekte im Wesentlichen von ihrer Entstehungsursache und Intensität ab und nur in einem sehr geringen Maße vom Nikotin selber.
Es gibt noch weitere Ansätze, Ursachen der Nikotin-Selbstmedikation zu analysieren. In einer an der University of Montana in den USA durchgeführten Studie, deren Ergebnisse im Jahre 2009 im Journal of American College Health erschienen, wurde die Rolle subjektiver Erwartungen in diesem Zusammenhang untersucht. Die klinische Psychologin Holly Schleicher fand mit ihrem Team heraus, dass die Erwartungen der Raucher an den positiven Effekt des Nikotins den Assoziationsgrad zwischen Tabakrauchen und einer Depression beeinflusst bzw. erhöht (Quelle: H. E. Schleicher, N. Nazir et al., The Role of Depression and Negative Affect Regulation Expectancies in Tabacco Smoking Among College Students, Department of Psychology, University of Montana, Missoula/MT, USA, Journal of American College Health, 57(5), S. 507 ‑ 512, Routledge, Abingdon-on-Thames, U. K., https:/www.ncbi.nlm.nih.gov/pubmed/...). In dieser Frage könnte daher auch die Psychologie eine Rolle spielen.
Im Gegensatz zum Ergebnis der oben zitierten Kendler-Studie kam David M. Fergusson mit seinem Team in einer an der University of Otago in Neuseeland durchgeführten langjährigen Studie zu dem Schluss, dass eine kausale Beziehung zwischen Rauchen als auslösendem Faktor und einer Depression wahrscheinlicher ist als der umgekehrte Zusammenhang. Die Studienergebnisse erschienen im Jahre 2010 im renomierten The British Journal of Psychiatry (Quelle: David M. Fergusson, Joseph M. Boden, L. Johne Horwood, Cigarette smoking and depression: tests of causal linkages using a longitudinal birth cohort, Department of Psychological Medicine, Christchurch Health and Development Study, University of Otago, Christchurch School of Medicine and Health Sciences, Christchurch, Neuseeland, The British Journal of Psychiatry, 196, S. 440 - 446, London/U. K. 2010, http://bjp.rcpsych.org/...). Einige Zeit vorher, im Jahre 2003, war in einer anderen Fachzeitschrift ein erster Artikel über das umfangreiche Projekt veröffentlich worden (Quelle: Fergusson, Goodwin, Horwood, Major depression and cigarette smoking: results of a 21-year longitudinal study, Psychological Medicine, 2003 (33), S. 1.357 ‑ 1.367, Cambridge Universitiy Press, Cambridge/U. K., https://www.cambridge.org/core/...).
Das neuseeländische Studienteam führte die Untersuchung mit einer Geburtskohorte von 1.265 jungen Erwachsenen durch, davon 635 Männer und 630 Frauen. Bis zu ihrem 16. Lebensjahr wurden die Teilnehmer jährlich kontrolliert und dann im Alter von 18, 21 und 25 Jahren befragt. Zur Auswertung wandten die Forscher ein lineares Paneldatenmodell und das Strukturgleichungsmodell (SEM) an, beides komplexe statistische Rechenmodelle.
Sie stellten zunächst fest, dass affektive Symptome innerhalb der Kohorte nikotinsüchtiger Probanden im Vergleich mit den nichtrauchenden Probanden mehr als doppelt so häufig vorkamen. Die Anwendung des SEM ergab dann eine höhere Wahrscheinlichkeit für die Nikotinsucht als auslösendem Faktor des Auftretens von Depressionssymptomen (Ergebnisparameter: 0,246 für die Wechselwirkung und 0,181 für den direkten Zusammenhang) im Vergleich mit der Wahrscheinlichkeit des umgekehrten Zusammenhangs, nämlich dass die Depression Ursache der Nikotinsucht ist (deutlich kleinere Parameterwerte: ‑0,025 für die Wechselwirkung und 0,047 für den direkten Zusammenhang).
Die Ergebnisse der Rechenmodelle liefern keine endgültigen Beweise für eine Kausalbeziehung, legen eine solche jedoch nahe. Die Studienergebnisse lassen auch auf die Existenz eines zweiten korrelativen Ursache-Wirkungs-Pfads mit gemeinsamen Risikofaktoren für Rauchen und Depression schließen.
Über die auslösenden Faktoren konnte die Studie keine Hinweise liefern, denn diese Fragestellung wurde nicht in die Untersuchung einbezogen. Die drei Autoren äußerten in ihrem Artikel jedoch die Möglichkeit, dass auf Nikotin basierende Neurotransmitteraktivitätsveränderungen eine wahrscheinliche neurobiologische Erklärung für den Kausalzusammenhang sind.
In derselben Ausgabe des The British Journal of Psychiatry, in der die Autoren der Fergusson-Studie im Jahre 2010 ihre Analysen vorstellten, veröffentlichten Marcus R. Munafò und Richardo Araya einen Beitrag, der sich mit den Möglichkeiten beschäftigt, den Beziehungscharakter zwischen Rauchen und Depression genauer zu ermitteln. Dabei setzten sie sich auch mit dem Vorgehen bei der Fergusson-Studie kritisch auseinander, beispielsweise die dort favorisierte Auswahl verschiedener Symptome einer Depression statt einer diagnostizierten Depression oder die verwendeten Nikotinabhängigkeitskriterien (Quelle: M. R. Munafò, R. Araya, Cigarette smoking and depression: a question of causation, The British Journal of Psychiatry, 196, S. 425 ‑ 426, London/U. K. 2010, http://www.bristol.ac.uk/...).
Munafò und Araya weisen auf die paradoxe Situation hin, die durch Erkenntnisse empirischer Studien entsteht, Rauchen könne eine Depression (mit-)verursachen, Raucher selber aber berichten, mit dem Rauchen negative Befindlichkeiten, wie Angst oder negative Stimmungen, positiv beeinflussen zu können. Eine mögliche Erklärung sehen sie in der kurzen Halbwertszeit des Nikotins einerseits und der Zeitverzögerung andererseits, mit der Entzugserscheinungen, wie Angstgefühle oder negative Affekte, erst viele Stunden nach dem letzten Tabakkonsum auftreten. Der durch das Rauchen unmittelbar ausgelöste Rückgang von Symptomen wird von den Tabakkonsumenten nämlich sofort und massiv wahrgenommen, während die Entzugserscheinungen hingegen von ihnen gar nicht mit Nikotin bzw. einem Entzug in Verbindung gebracht werden.
Die Autoren sehen den Beweis für einen direkten Zusammenhang zwischen Rauchen und einer Depression durch die Erfahrungen mit den Ergebnissen erfolgreicher Nikotinentzugstherapien erbracht, bei denen sich die Stimmung der Konsumenten deutlich verbessert.
Da Beobachtungsstudien Kausalitäten auch mit noch so ausgefeilten Statistikmodellen nicht vollständig beweisen können und experimentelle Studien aus ethischen Gründen abgelehnt werden, schlagen die beiden Autoren die Anwendung der Methode der Mendelschen Randomisierung vor, um die Frage der Kausalität von Tabakkonsum und Depression besser beantworten zu können.
Die von Munafò und Araya vorgetragene Begründung für einen direkten Kausalzusammenhang zwischen Tabakkonsum und einer Depression, Rauchstopp führe zu einer deutlichen Verbesserung der Stimmung, konnte in verschiedenen Studien nachgewiesen werden.
Im Jahre 2010 wurden die Ergebnisse einer von dem klinischen Psychologen Christopher W. Kahler geleiteten Studie veröffentlicht, die den von Munafò und Araya beschriebenen Zusammenhang nahelegen (Quelle: Christopher W. Kahler, Adam M. Leventhal et al., Time-Varying Smoking Abstinence Predicts Lower Depressive Symptoms Following Smoking Cessation Treatment, Center for Alcohol and Addiction Studies and Centers for Behavioral and Preventive Medicine/The Miriam Hospital, Brown University, Providence/Rhode Island, USA, University of Southern California, Keck School of Medicine, Los Angeles/CA, USA, Nicotine & Tobacco Research, Oxford University Press, Oxford/U. K. 2010, https://pmalpha.usc.edu/...).
Es wurden die Reaktionen von 236 alkoholabhängigen Männern und Frauen, überwiegend mit weißer Hautfarbe und nicht hispanischen Ursprungs, auf einen Rauchstopp getestet. Dabei wurden sie auf Symptome einer Affektiven Störung eine Woche vor dem Entzugsbeginn und anschließend vier Mal ‑ bis zur 28. Woche nach Beginn ‑ untersucht. Ein kleiner Teil der Gruppe von 33 Personen blieb auch nach Studienende abstinent, während der Rest es entweder nur kurzzeitig schaffte, rauchfrei zu bleiben, oder sogar ohne Unterbrechung weiter rauchte.
Die Ergebnisse ließen an Eindeutigkeit nichts zu wünschen übrig. Die erfolgreichen Personen aus der Gruppe der 33 abstinenten ehemaligen Raucher wiesen nach dem Rauchstopp kaum Symptome einer Depression bzw. depressiven Verstimmung auf. Die Stimmung verschlechterte sich zunehmend, je erfolgloser ein Teilnehmer war. Personen, die es zumindest zeitweise schafften, rauchfrei zu bleiben, hatten in dieser Zeit eine wesentliche bessere Stimmung als in der übrigen Zeit, in der ihre Stimmung teilweise sogar unter das Niveau sank, das sie zu Studienbeginn aufwiesen. Konstant weiter rauchende Teilnehmer hatten die schlechteste Stimmung mit den meisten Symptomen einer Depression.
Auch in dieser Studie konnten über die exakten Auslöser der Stimmungslagenveränderungen, das heißt ob psychologische, neurobiologische oder ein Mix aus beidem, keine Aussagen getroffen werden, da diese Thematik nicht untersucht wurde.
Die Auswahl des Personenkreises deutet zunächst darauf hin, dass die Ergebnisse nicht 100%ig auf die Gesamtbevölkerung übertragen werden dürfen. Allerdings kann Kahler auf ähnliche Resultate in einer von ihm im Jahre 2002 durchgeführten Studie mit einer repräsentativeren Teilnehmerschaft verweisen (Quelle: C. W. Kahler et al., Negative mood, depressive symptoms, and major depression after smoking cessation treatment in smokers with a history of major deressive disorder, Journal of Abnormal Psychology, 111, 11/2002, American Psychological Association, Washington, D.C, USA, http://psycnet.apa.org/...) und kommt daher zu dem Schluss, dass auch die Ergebnisse der neuen Studie generalisiert werden dürfen. Im Studiendokument wird darüber hinaus auf ähnliche Untersuchungen anderer Wissenschaftler mit vergleichbaren Ergebnissen hingewiesen: S. Cohen, E. Lichtenstein 1990, J. R. Hughes 1992, A. C. Parrot 1995, R. West, P. Hajek 1997, L. Chassin, K. Kim et al. 2002 und M. R. Munafò, R. Araya et al. 2008.
Studienleiter Christopher Kahler fasst die Ergebnisse der Untersuchung knapp zusammen: „If they quit smoking their depressive symptoms go down and if they relapse, their mood goes back to where they were. An effective antidepressant should look like that.“ (Quelle: Pressemitteilung der Brown University, Kicking the habit: Study suggests that quitting smoking improves mood, ScienceDaily, Rockville/MD, USA, 12/2010, https://www.sciencedaily.com/...).
Im Jahre 2011 bildete eine groß angelegte Befragung, die in Kanada zwischen den Jahren 1994 und 2007 durchgeführt wurde, der Psychiaterin Salma M. Khaled und einem Team aus mehreren kanadischen Universitäten die Grundlage für die Untersuchung des Zusammenhangs zwischen intensivem Rauchen und einer Depression (Quelle: Salma M. Khaled, Andrew G. Bulloch, Scott B. Patten et al., Persistent heavy smoking as risk factor for major depression (MD) incidence ‑ Evidence from a longitudinal Canadian cohort of the National Population Health Survey, Mental Health Center for Research and Teaching, Toronto/Canada, University of Calgary, Calgary/Canada, Hotchkiss Brain Institute, Calgary/Canada, Journal of Psychiatric Research, 46/2012, S. 436 ‑ 443, http://www.journalofpsychiatricresearch.com/...).
Vergleichbar mit der Fergusson-Studie, die ein Jahr zuvor veröffentlicht wurde, stützen die Resultate auch hier die Vermutung eines direkten Kausalzusammenhangs. Die Khaled-Studie basiert auf einer noch größeren Datenmenge im Vergleich mit der Fergusson-Studie, nämlich 3.824 (statt 1.265) Teilnehmer. Außerdem wird die eindeutige Diagnose einer Major Depression als Merkmal bevorzugt und nicht einzelne Depressionssymptome. Eine Tabakabhängigkeit wird ab einem Konsum von 20 Zigaretten pro Tag eindeutig festgelegt.
Für den gesamten Zeitraum der Erhebung von 1994 bis 2007 lag die Wahrscheinlichkeit der Diagnose einer Major Depression in der Gesamtkohorte bei 13,2%. Bei starken Rauchern erhöhte sich das Risiko gegenüber Ex-Rauchern von 7,1% auf 26,7% um mehr als das Dreifache. Verglichen mit den starken Rauchern nahm das Erkrankungsrisiko bei Ex-Rauchern von dem Zeitpunkt an kontinuierlich ab, an dem sie das Rauchen beendeten. Das Risikoverhältnis sank um 0,5 nach einer Abstinenzzeit von einem bis fünf Jahren und auf 0,2 für langjährige Abstinenzler ab dem 21. rauchfreien Jahr.
Khaled sieht den kausalen Zusammenhang durch ihre Untersuchungen bestätigt und argumentiert plausibel mit dem Umstand, dass das Risiko der Anfälligkeit für eine Depression bei derzeitigen und ehemaligen Rauchern gleich oder annähernd gleich sein müsste, wenn beide Merkmale ausschließlich auf einer gemeinsamen Ursache beruhen, das heißt nur korrelativ miteinander verbunden sind. Die außerordentlich hohen Abweichungen zwischen beiden Gruppen deuten nicht auf eine Korrelation hin.
Neben der Kausalitätsfrage wird empirisch auch die Frage der Nachhaltigkeit von Nikotinschäden untersucht: Handelt es sich bei den hirnorganischen Veränderungen um kurzfristige, sich bei Abstinenz zurückbildende, oder eher um langfristige Anpassungen, und welche Auswirkungen könnte dies auf das Risiko haben, an einer Affektiven Störung zu erkranken?
Wie weiter oben schon erwähnt, wird in der Forschung eher von einer vollständigen Rückbildung der Veränderungen nicotinischer Acethylcholin-Rezeptoren bei kosequenter Tabakabstinenz aus. Es gibt aber keine absolute Gewissheit in dieser Frage und darüber hinaus auch andere Rezeptortypen, die mit Nikotin interagieren. Wären Schäden ganz oder teilweise langfristig, ist auch von langfristigen Auswirkungen auf die Höhe des Erkrankungsrisiko auszugehen. Einige Untersuchungen weisen genau auf diesen Umstand hin.
Im Januar 2013 erschienen in der Zeitschrift PNAS die Ergebnisse einer Studie dreier Universitäten aus der Schweiz unter der Leitung des Berner Psychiaters Gregor Hasler, in der sich mit den langfristigen Auswirkungen des Nikotins auf das Glutamat-System auseinandergesetzt wurde (Quelle: Funda Akkus, Simon M. Ametamey, Gregor Hasler et al., Marked global reduction in mGluR5 receptor binding in smokers and ex-smokers determined by [11C]ABP688 positron emission tomography, Procceedings of the National Academy of Sciences, Washington D. C./USA 2013, doi: 10.1073/pnas.1210984110, https://www.ncbi.nlm.nih.gov/...).
Die Untersuchung ergab, dass durch Nikotin verursachte hirnorganische Veränderungen den Ex-Rauchern auch nach einer längeren Abstinenz zu schaffen machen können. Die Gehirne von Rauchern, Ex-Rauchern und Nichtrauchern wurden mit der PET-Technologie visualisiert. Dabei wiesen Raucher zunächst eine deutliche Verringerung der Anzahl des GluR5-Rezeptortyps von 20 bis 30% gegenüber Nichtrauchern auf, die man in dieser Stärke nicht erwartet hatte. Ex-Raucher wiesen nach einer sechsmonatigen Abstinenz immer noch eine Reduktion der mGluR5-Rezptoren um 10 bis 20% auf. „«Diese Veränderung des Glutamat-Systems bei Rauchern ist im Ausmaß und in der lokalen Ausweitung weit größer, als man bisher angenommen hat», erläutert Hasler. Besonders unerwaretet sei, dass die Erholung des Glutamat-Systems offenbar sehr lange dauere. «Es ist wahrscheinlich, dass diese sehr langsame Normalisierung zu der sehr hohen Rückfallquote bei Ex-Rauchern beiträgt.»“ (Quelle: www.scinexx.de, Rauchen beeinflusst das Gehirn nachhaltig, MMCD NEW MEDIA, Düsseldorf 2012, http://www.scinexx.de/...).
Vier Forscherinnen der University of California um die Pharmazeutin Menglu Yuan veröffentlichten im Jahre 2015 eine Analyse und Zusammenfassung der Ergebnisse zahlreicher anderer Studien, in denen man sich mit den Auswirkungen des Tabaks und Nikotins auf das jugendliche Gehirn beschäftigt hatte (Quelle: Menglu Yuan, Sarah J. Cross, Sandra E. Loughlin, Frances M. Leslie, Nicotine and the adolescent brain, School of Medicine, University of California, Irvine/CA, USA, The Journal of Physiology, 593 (16), S. 3397 ‑ 3412, The Physiological Society, Wiley-Blackwell, London/U. K., 2015, doi: 10.1113/JP270492, http://www.trdrp.org/files/...).
Sie zeigen mit ihrer Literaturstudie, dass die Wirkungen von Tabak und Nikotin bei Jugendlichen nicht zu vergleichen sind mit denen bei Erwachsenen. Das jugendliche Gehirn reagiere empfindlicher und langfristiger auf eine Nikotinbelastung. Außerdem erhöhe sich durch Tabakkonsum die Empfänglichkeit der Jugendlichen für andere Drogen, beispielsweise für Kokain.
Betrachtet man noch einmal Abbildung 40 über die Entwicklung des Gehirns in Kindheit und Jugend (→ Abschnitt 4.12.4), so wird unmittelbar verständlich, warum der schädliche Einfluss des Nikotins zwangsläufig langfristig sein muss.
Aufgrund der Komplexität der Auf- und Umbaumaßnahmen, von denen das jugendliche Gehirn bis etwa zum 24. Lebensjahr betroffen ist und die bis heute nicht vollständig verstanden werden, ist es jedoch schwierig, Nikotinwirkungen detailiert zu bestimmen und zu bewerten. Es ist der Arbeit von Menglu Yuan und ihrem Team zu verdanken, etwas mehr Transparenz in diese komplizierte Angelegenheit gebracht zu haben.
Hier nur einige der wichtigsten Ergebnisse, die im Abschlussdokument zitiert werden und einen Bezug zu Affekten und Affektiven Störungen haben:
- 90% der erwachsenen Raucher beginnen ihren Konsum vor dem 18. Lebensjahr (Quelle: SAMHSA, U. S. Department of Health and Human Services, Rockville/MD, USA).
- Die Wahrscheinlichkeit, langfristig Tabak zu konsumieren, ist gering, wenn nicht schon in der Adoleszenz-Phase mit dem Rauchen angefangen wurde (Quelle: S. Sussmann, 2002, Effects of sixty six adolescent tobacco use cessation trials and seventeen prospective studies of self-initiated quitting, Tobacco Induced Diseases, International Society for the Prevention of Tobacco Induced Diseases, Heraklion/Griechenland). Fast alle Erwachsenen haben damit ihre „Raucherkarriere“ in ihrer Jugendzeit begonnen.
- Jugendliche Raucher erkranken häufiger an psychiatrischen Störungen als Nichtraucher (Quellen: M. McKenzie M, C. A. Olsson et al., 2010, Association of adolescent symptoms of depression and anxiety with daily smoking and nicotine dependence in young adulthood: findings from a 10-year longitudinal
study, Addiction, 105, Wiley-Blackwell, Hoboken/NJ, USA und S. H. Kollins, R. A. Adcock, 2014, ADHD, altered dopamine neurotransmission, and disrupted reinforcement processes: implications for smoking and nicotine dependence, Progress in Neuro-Psychopharmacology & Biological Psychiatry, 7/2014, Elsevier, London/U. K., 2014).
- Tierversuchsstudien zeigten, dass sich die in der Adoleszenz entwickelnden Hirnareale, unter anderem die für das emotionale Erleben, als sehr anfällig gegenüber einer Exposition durch Nikotin erwiesen haben. Das gilt unabhängig von Fristigkeit und Dosierung. Auch geringe und kurzfristig verabreichte Mengen Nikotins sind schon in der Lage, ungünstige Veränderungen in diesen Gehirnregionen zu provozieren (Quelle: S. D. Iñiguez et al., Nicotine exposure during adolescence induces a depression-like state in adulthood, Neuropsychopharmacology, 34, Nature Publishing Group, London/U. K, 2009).
- Auch in der Kausalitätsfrage kamen die Forscherinnen zu einem interessanten Ergebnis, denn dass Tabakkonsum Jugendlicher nicht nur in einem einfachen Zusammenhang mit negativen Veränderungen der Stimmungslage steht, sondern diese auch induzieren kann, wird in mehreren Studien festgestellt (Quellen: John et al., Smoking,
nicotine dependence and psychiatric comorbidity ‑ a population-based study including smoking cessation after three years, Drug and Alcohol Dependence, 76 (3), Elsevier, Amsterdam/NL, 2004; O. Klungsoyr et al., Cigarette smoking and incidence of first depressive episode: an 11‑year, population-based follow-up study, American Journal of Epidemiology, 163, Oxford University Press/Johns Hopkins Bloomberg School of Public Health, Baltimore/MD, USA, 2006; T. Flensborg-Madsen et al., Tobacco smoking as a risk factor for depression. A 26-year population-based follow-up study, Journal of Psychiatric Research, 45 (2), Elsevier B. V., Amsterdam/NL, 2011).
Zwei der wichtigsten Aspekte der Gefahren des Tabakkonsums während der Adoleszenz, auf die in der Studie hingewiesen wird, sind die langfristigen neurobiologischen Veränderungen des Gehirns, beispielsweise durch den störenden Einfluss des Nikotins auf die Reifung nicotinischer Rezeptoren, und die damit einhergehenden langfristigen Veränderungen im Suchtverhalten, bei kognitiven Fähigkeiten und in der Regulation von Affekten.
Das Einstiegsalter wirkt sich erheblich aus: Je früher mit dem Tabakrauchen begonnen wird, desto gravierender sind die negativen Entwicklungen im Zentralnervensystem. Es ist damit ein Unterschied, ob im 14. Lebensjahr, im 18. Lebensjahr oder im 21. Lebensjahr mit dem Rauchen begonnen wird.
Rauchen während der Schwangerschaftsphase stellt eine erhebliche Gefahr für die Entwicklung des Fötus dar, insbesondere für dessen sich entwickelndes fetales Gehirn, mit gravierenden Auswirkungen bis ins Erwachsenenalter.
Ein im Jahre 2008 in der Zeitschrift Der Nervenarzt erschienener Artikel von Carina Wessels und Georg Winterer, der noch heute relevant ist, bietet einen ausführlichen Überblick zu diesem Thema, auch wenn sich in der Zwischenzeit auf diesem Forschungsgebiet wahrscheinlich eine Menge weiterer Erkenntnisse ergeben haben (Quelle: Carina Wessels, G. Winterer, Nikotin und Gehirnentwicklung, Psychiatrische Klinik der Heinrich-Heine-Universität, Düsseldorf, Der Nervenarzt, 2/2008, Springer Medizin, Heidelberg/Neu-Isenburg 2008, http://link.springer.com/...).
In der folgenden Zusammenfassung des Beitrags werden nicht alle Studien benannt, aus denen Wessels und Winterer zitieren. Der o. g. Link führt direkt zu einer Seite mit der Möglichkeit, ein Pdf des Artikels herunterzuladen; dort findet man die komplette Liste mit allen Quellenangaben.
Zunächst weisen die Autoren auf den Umstand hin, dass 25% aller schwangeren Frauen in Deutschland rauchen (Quelle: Ali H. Bardy, Timo Seppala, Pirjo Lillsunde et al., 1993, Objectively measured tobacco exposure during pregnancy: neonatal effects and relation to maternal smoking, British Jorunal of Obstetrics and Gynaecology, 100, 8/1993, Wiley-Blackwell, Oxford/U. K., http://onlinelibrary.wiley.com/...).
Auch wenn es in diesem Abschnitt hauptsächlich um die direkten neurologischen Implikationen des Tabak- und Nikotinkonsums geht, soll der folgende wichtige Umstand nicht unerwähnt bleiben, denn er hat einen großen und negativen Einfluss auf die Entwicklung des Nervensystems des Kindes dar. Ein Problem stellen nämlich tabakbedingte Schädigungen der Placenta und dadurch bedingte Störungen des Nährstoff- und Sauerstoffaustauschs zwischen Mutter und Fötus dar, die nachgewiesenermaßen postnatal bei 50% der Kinder zu Hirnanomalien und bei 28% zu cerebralen Blutungen führen.
Auf der fetalen Plazentaseite ist die Anzahl der Nikotinrezeptoren bei rauchenden Müttern um ca. 15% höher. Kurz nach der Geburt leiden Säuglinge, deren Mütter geraucht haben, deshalb für ca. zwei Tage an einem Nikotinentzugssyndrom. Die beiden Autoren führen insgesamt sieben Studien auf, die auf diese Umstände hinweisen.
Als noch schwerwiegender werden die nikotinbedingten neurologischen Auswirkungen auf verschiedene entstehende Neurotransmittersysteme und die Neuronenverschaltung eingeschätzt und durch drei Quellen belegt. Dadurch könne es zu einer veränderten Nikotinsensibilisierung des Gehirns kommen (Quelle: Xiaobin Wang, Barry Zuckerman, Colleen Pearson et al., 2002, Maternal Cigarette Smoking, Metabolic Gene Polymorphism, and Infant Birth Weight, Journal of the American Medical Association, 287 (2), 1/2002, http://www.ipr.northwestern.edu/...) mit Folgen für das spätere Leben. Mit drei weiteren Quellen wird dargelegt, dass dadurch manifeste Lernschwierigkeiten, Änderungen des Verhaltens, Aufmerksamkeitsdefizite, Hyperaktivität oder eine Neigung zur Nikotinsucht die Folgen sein können.
Der Zusammenhang von mütterlichem Tabakkonsum und der Entstehung eines ADHS-Syndroms ist mittlerweile gut belegt. Eine Zwillingsstudie ergab starke Anhaltspunkte für einen kausalen Zusammenhang (mehrere Quellen, u. a. Sharon Milberger, Joseph Biederman, Stephen V. Faraone et al., 1996, Is Maternal Smoking During Pregnancy a Risk Factor for Attention Deficit Hyperactivity Disorder in Children?, The American Journal of Psychiatry, 153, 9/1996, American Psychiatric Association, Arlington County/VA, USA, http://ajp.psychiatryonline.org/... und Anita Thapar, Tom Fowler, Frances Rice et al., 2003, Maternal Smoking During Pregnancy and Attention Deficit Hyperactivity Disorder Symptoms in Offspring, The American Journal of Psychiatry, 160, 11/2003, American Psychiatric Association, Arlington County/VA, USA, https://ils.unc.edu/...).
Wessels und Winterer verweisen auf mehr als zehn Tierversuchsstudien, die sich mit neurobiologisch-pränatalen Veränderungen nach einer Nikotinexposition beschäftigen und massive Hirnschädigungen und Veränderungen am Dopamin-, Serotonin- und Acetylcholinstoffwechsel zeigten, letztere mit den Folgen einer vorzeitigen Beendigung der fetalen Gehirnentwicklung (Quellen: H. A. Navarro, F. J. Seidler, R. D. Schwartz et al., 1989, Prenatal Exposure to Nicotine Impairs Nervous System Development at a Dose Which Does Not Affect Viability or Growth, Brain Research Bulletin, 9/1989, 23 (3), Elsevier B. V., Amsterdam/NL, http://ac.els-cdn.com/... und William Slikker Jr., Z. Alex Xu, Edward D. Levin, Theodore A. Slotkin, 2005, Mode of Action: Disruption of Brain Cell Replication, Second Messenger, and Neurotransmitter Systems During Development Leading to Cognitive Dysfunction-Developmental Neurotoxicity of Nicotine, Critical Reviews in Toxicology, 2005, 35 (8/9), Taylor & Francis, Milton Park, Abingdon-on-Thames/U. K, http://www.tandfonline.com/...).
Alle genannten Indizien bestätigen die Plausibilität der kausaltheoretischen Sichtweise, dass Nikotin- und Tabakkonsum während der Schwangerschaft eine Noxe darstellen, die mit hoher Wahrscheinlichkeit beim Nachwuchs u. a. später auch die Entstehung einer Affektstörung begünstigen kann. Die Frage ist nun: Welche Erkenntnisse hat die empirische neurobiologische Forschung hier zu bieten? Leider findet man in dem zitierten Beitrag von Wessels und Winterer zu diesem Thema keine Informationen.
Im Jahre 1998 veröffentlichten David M. Fergusson und ein Team der Christchurch School fo Medicine die Ergebnisse einer mit 1.265 Kindern bzw. Jugendlichen in Neuseeland durchgeführten 18-Jahres-Langzeitstudie über die Auswirkungen des Rauchens während der Schwangerschaft auf die spätere psychiatrische Gesundheit dieser Jugendlichen zwischen ihrem 16. und 18. Lebensjahr (Quelle: David M. Fergusson, Lianne J. Woodward, John Horwood, Maternal Smoking During Pregnancy and Psychiatric Adjustment in Late Adolescence, Department of Psychological Medicine, Christchurch School of Medcine, Christchurch/Neuseeland, American Medical Association, Archives of General Psychiatry, 8/1998, 55, S. 721 ‑ 727, Chicago/USA, http://jamanetwork.com/...).
In ihrer Einführung weisen die Autoren darauf hin, dass die Ergebnisse mehrerer früherer Studien den Schluss zulassen, dass das Rauchen während der Schwangerschaft mit erheblichen ‑ teilweise langfristigen ‑ Risiken assoziert ist, unter anderem Fehlgeburt, reduziertes Geburtsgewicht, Intelligenzminderung, ADHS oder Verhaltensstörungen, während es über die meisten Zusammenhänge, die in dieser Studie Gegenstände der Untersuchung waren, bisher keine Erhebungen gegeben hätte, nämlich zu auffälligem Sozialverhalten, Erkrankungen an Depression oder generalisierten Angststörungen und Suchterkrankungen.
Die Ergebnisse der Studie deuten auf einen signifikanten Zusammenhang zwischen Rauchen während der Schwangerschaft und dem Auftreten von einigen dieser vier Störungen, beschränkt auf die Lebensspanne zwischen dem 16. und 18. Lebensjahr. Dabei wurden andere Risikofaktoren aus dem familiären und sonstigen Umfeld berücksichtigt, beispielsweise das Bildungsniveau oder sonstiger Suchtstoffkonsum der Mutter, um das Ergebnis statistisch von diesen Einflussfaktoren zu bereinigen.
Sowohl unbereinigt als auch bereinigt ergab sich ein deutlicher Zusammenhang zwischen dem Rauchen während der Schwangerschaft und dem Entstehen auffälligen Sozialverhaltens und Suchtverhalten. Der Zusammenhang mit einer späteren Depression war weniger stark ausgeprägt, während bei den Angststörungen keine Zusammenhänge festgestellt wurden.
Die Ergebnisse für die Kinder von Müttern, die während der Schwangerschaft mindestens eine Packung am Tag, das heißt 20 Zigaretten, rauchten, betrugen als Durchschnittsrate (mean rate) im Vergleich mit nichtrauchenden Müttern...
- für auffälliges Sozialverhalten unbereinigt 2,58 bzw. bereinigt 2,06,
- für Alkoholmissbrauch bzw. -abhängigkeit unbereinigt 1,81 bzw. bereinigt 1,44,
- für Nikotinabhängigkeit unbereinigt 2,02 bzw. bereinigt 1,19,
- für Missbrauch oder Abhängigkeit von sonstigen Substanzen unbereinigt 2,38 bzw. bereinigt 1,62,
- für eine Major Depression unbereinigt 1,43 bzw. bereinigt 1,18 und
- für eine generalisierte Angststörung 0,92 bzw. bereinigt 1,07.
Als Medianwert wird 2,0 bei den unbereinigten Ergebnissen angegeben.
Interessant war ein Geschlechtervergleich der Ergebnisse des Einflusses auf das Sozialverhalten. So ergab sich eine erhebliche Diskrepanz zwischen männlichen Jugendlichen (bereinigt 2,75) und weiblichen Jugendlichen (bereinigt 1,4). Leider liegen vergleichbare Zahlen für die restlichen drei Merkmale nicht vor.
Ein Jahr später, im Juli 1999, erschienen in den USA die Ergebnisse einer Studie mit ähnlicher Fragestellung, die unter der Leitung von Myrna Weissmann durchgeführt wurde (Quelle: Myrna M. Weissmann, Virginia Warner et al., Maternal Smoking During Pregnancy and Psychopathology in Offspring Followed to Adulthood, New York State Psychiatric Institute, New York/USA, Journal of the American Academy of Child and Adolescent Psychiatry, 38 (7), Juli 1999, Elsevier B. V., Amsterdam/NL, https://www.researchgate.net/publication/...). Dabei wurden nichtrauchende Mütter mit Müttern verglichen, die in der Schwangerschaft mindestens zehn Zigaretten täglich geraucht hatten.
Die Ergebnisse dieser Studie wies teilweise Parallelen mit der vorigen auf. Einen besonders starken Zusammenhang ergab sich für sozialverhaltensgestörte männliche Jugendliche bis zum 13. Lebensjahr. Dort betrug die Rate 4,1, das heißt das Risiko war gut vierfach erhöht gegenüber dem Risiko von Jugendlichen, deren Mütter während der Schwangerschaft nicht rauchten. Weibliche Jugendliche zwischen dem 13. und 17. Lebensjahr waren nach dieser Studie erheblich drogengefährdet mit dem mehr als dem 5fachen Risiko (Rate 5.36).
Für das hier zu diskutierende Thema sind die Ergebnisse zum Merkmal der Major Depression Desease (MDD) relevant. Hier konnte nur für männliche Jugendliche zwischen dem 13. und 17. Lebensjahr ein nennenswertes Risiko (Rate 1,71) festgestellt werden. Bei allen anderen Gruppen männlicher und weiblicher Nachkommen aller untersuchter Altersklassen konnte keine Zusammenhänge festgestellt werden.
Eingeschränkt wird die Aussagekraft beider Studien durch den Fokus auf die Gruppe der Kinder und Jugendlichen und dort auf bestimmte Altersklassen. Daher sind die Resultate auch nur für vergleichbare Gruppen aussagefähig, nicht jedoch für erwachsene Personen.
Welches Fazit ist aus allen ‑ theoretischen wie empirischen ‑ Erkenntnissen über das Verhältnis der Nikotinsucht zu Affektstörungen zu ziehen?
Die nachgewiesenen Suchtmechanismen demonstrieren auf eindeutige Weise, dass sich nikotinbedingte Veränderungen in affektrelevanten Hirnregionen manifestieren (→ Abbildung 47). Es besteht daher eine hohe Wahrscheinlichkeit, dass Nikotinkonsum eine Affektstörung begünstigt, es mithin einen kausalen Zusammenhang geben kann.
Kausaltheoretisch ist der Zusammenhang damit nachvollziehbar begründbar. Aber auch emprirische Untersuchungen bestätigen diese Sicht und stützen damit die in dieser Publikation vertretene Noxen-Theorie.
Das bedeutet jedoch nicht, dass ein umgekehrter Kausalzusammenhang ausgeschlossen ist, und eine Affektstörung den Bedarf nach Tabakkonsum triggern kann.
Ebenfalls bedeutet das nicht, dass die Möglichkeit einer Korrelation - das heißt einer gemeinsamen Ursache für beide Merkmale - verneint wird. Auch einen solchen Zusammenhang legen empirische Studien nahe.
Eine Kombination dieser unterschiedlichen Zusammenhangs-Szenarien und deren Ineinandergreifen ist sehr wahrscheinlich und lässt sich auch anhand des in Abbildung 48 dargestellten Kreislaufmodells erklären.
Ob nikotinbedingte Veränderungen ganz oder teilweise einen chronischen Verlauf nehmen oder sich kurz- bis mittelfristig zurückbilden, konnte durch Studien noch nicht ausreichend belegt werden. Es gibt zumindest Hinweise, dass es zu langfristigen Schäden im Gehirn kommen kann.
Einen deutlichen Einfluss hat das Einstiegsalter. Je jünger ein Mensch beim Beginn des regelmäßigen Tabakkonsums ist, desto nachhaltiger sind die nikotinbedingten neurologischen Veränderungen und desto wahrscheinlicher ist, dass sich diese Veränderungen langfristig auch auf den psychiatrischen Zustand auswirken und damit die Wahrscheinlichkeit erhöhen, im Laufe des Lebens an einer Affektstörung zu erkranken.
Demgegenüber konnte empirisch noch kein signifikanter Zusammenhang zwischen dem Rauchen während der Schwangerschaft und einer späteren Affektstörung jugendlicher Nachkommen nachgewiesen werden. Es wurden lediglich leicht erhöhte Risiken für die Betroffenen festgestellt. Die Ergebnisse beruhen allerdings auf nur wenigen Untersuchungen und betreffen auch nicht die Lebenszeitprävalenz, sondern nur die Jugendzeit.
Alkohol und Alkoholabhängigkeit
Bei der Diskussion der schädlichen Auswirkungen des Alkohols werden grundsätzlich Alkoholmissbrauch und Alkoholabhängigkeit unterschieden. Die internationalen statistischen Klassifikationssystemen für Erkrankungen bzw. psychische Störungen, ICD‑10 und DSM, führen beide Formen getrennt auf.
Hier ist diese Differenzierung nur insofern von Bedeutung, da die Alkoholabhängigkeit als eine mit einem chronisch starken Verlangen nach Alkoholkonsum und mit Entzugserscheinungen einhergehende Erkrankung assoziiert wird und damit neurobiologische Ursachen nahelegt. Im Gegensatz dazu weist der Alkoholmissbrauch per Definition keine derartigen Merkmale auf, obwohl es nach gängiger Meinung auch hier zu neurologischen Störungen bzw. Schäden kommen kann. Diese Unterscheidung ändert auch nichts daran, dass Alkoholmissbrauch häufig eine Vorstufe der Alkoholabhängigkeit darstellt. So ist zum Beispiel das Rauschtrinken Jugendlicher eine Form des Alkoholmissbrauchs und kann später zur Alkoholabhängigkeit beitragen.
Im Folgenden stehen - wie schon beim Nikotin - die direkten zentralnervösen neurobiologischen Auswirkungen des Alkohols und deren Folgen hinsichtlich affektiver Ekrankungen im Vordergrund. Ein theoretischer und/oder empirischer Nachweis kausaler Zusammenhänge zwischen Alkohol bzw. Alkoholismus und affektiven Erkrankungen würde Alkohol im Rahmen einer multikausalen Theorie als Noxe bestätigen.
Wie die meisten Suchtstoffe hat Alkohol zusätzlich noch das Potential, das zentrale Nervensystem indirekt zu schädigen, beispielsweise durch Gefäß- oder Leberschäden oder auch durch fehlende neurologisch bedeutsame Mikronährstoffe (beispielsweise Thiamin/Vitamin B1), basierend auf alkoholbedingtem Ernährungsfehlverhalten oder alkoholbedingten Störungen des Verdauungssystems. Ungünstige Auswirkungen auf das Gefäßsystem könnten auch ein Grund für pathologische Veränderungen der Blut-Hirn-Schranke sein, die viele negativen Folgen für das Gehirn haben können.
Indirekte Alkoholauswirkungen auf Zentralnervensystem und Affekt werden in Abschnitten über die jeweilige Primärursache thematisiert (→ zum Beispiel Vaskuläre Erkrankungen im Abschnitt 4.14 oder Mikronährstoffmangel und Affektstörungen im Abschnitt 4.4). Sie spielen an dieser Stelle nur am Rande eine Rolle.
Neben zentralnervösen Auswirkungen besteht ebenfalls der Verdacht, dass auch das periphere Nervensystem, beispielsweise der parasympatische Nervus vagus, alkoholbedingt Schaden nimmt.
Neurologisch gesehen hat Alkohol eine Menge direkter negativer zelltoxischer und psychoaktiver Wirkungen. Eine Übersicht über die Konsequenzen psychoaktiver Alkoholaktivitäten für verschiedene Transmissionssyteme mit Verweisen auf entsprechende Studien findet man in der Schweizer Zeitschrift Psychiatrie & Neurologie, die nachfolgend zusammengefasst dargestellt wird (Quelle: Lorenz Deutschbaur, Marc Walter, Neurobiologische Effekte von Alkohol, Psychiatrie & Neurologie, 1/2014, Rosenfluh Publikationen AG, Neuhausen/Schweiz 2014, http://www.tellmed.ch/tellmed/...).
Im opiodergen Transmittersystem führt Alkoholkonsum u. a. zu einer Vermehrung der Anzahl des μ‑Opioidrezeptors sowohl im Nucleus accumbens der Basalganglien als auch im orbifrontalen Cortex. Im Falle des Alkoholkonsums steigt die Ausschüttung von Endorphinen, die nun auf eine vermehrte Anzahl von μ‑Opioidrezeptoren treffen und eine verstärkte Ausschüttung Dopamins in beiden Hirnregionen bewirken.
Im serotonergen Transmittersystem bewirkt Alkohol eine Erhöhung der Serotoninsynthese, woraufhin der Serotoninspiegel im gesamten Gehirn steigt. Im Nucleus accumbens wird die Dopaminausschüttung durch Serotonin moduliert, da die dortigen Nervenzellen über serotonerge 5‑HT3‑Rezeptoren verfügen. Ein durch Alkohol zentralnervös erhöhter Serotoninspiegel führt damit im Nucleus accumbens zu einer erhöhten Menge freigesetzten Dopamins. Das scheint aber nur für Nicht-Alkoholiker zu gelten, denn verschiedene Studien ergaben eine sinkende Dopaminproduktion bei alkoholabhängigen Menschen.
Im GABA-nergen Transmittersystem aktiviert Alkohol die GABA‑A‑Rezeptoren. Dadurch wird die Durchlässigkeit der Zellmembran für Chloridionen erhöht mit dämpfenden Effekten auf die Zellerregung. GABA‑A‑Rezeptoren kommen im Gehirn häufig vor, insbesondere im Hippocampus und in der gesamten Großhirnrinde. Bei alkoholabhängigen Menschen ist die Anzahl der GABA‑A‑Rezeptoren reduziert.
Im glutamatergen Transmittersystem wird durch Alkohol der NMDA-Rezeptor blockiert, der eigentlich für Glutamat gedacht ist und damit dessen Übertragung verhindert. Glutamat ist der wichtigste erregende Botenstoff im Zentralnervensystem. Bei chronischem Alkoholkonsum steigt als Gegenreaktion die Anzahl der NMDA-Rezeptoren. Darüber hinaus beeinflusst Alkohol im glutamatergen Transmittersystem auch die AMPA-Rezeptoren mit vergleichbaren Folgen für die Übertragung von Glutamat. NMDA-Rezeptoren findet man im gesamten Großhirn und vor allem im Hippocampus, AMPA-Rezeptoren sind im gesamten Gehirn im Vergleich mit anderen Rezeptoren am häufigsten vertreten. In bestimmten Hirnregionen, beispielsweise im Hippocampus, wird die Anzahl der AMPA-Rezeptoren an die Aktivität der Synapse angepasst. Sinkt alkoholbedingt die Synapsenaktivität aufgrund der Glutamatblockade durch Alkohol, erhöht sich dort automatisch die Anzahl der AMPA-Rezeptoren.
Über die Wirkungen des Alkohols auf das dopaminerge Transmittersystem schreiben Deutschbaur und Walter: „Gute Daten liegen für eine alkoholinduzierte Zunahme der dopaminergen Transmission im mesolimbischen System vor. Bei Laborratten stimuliert akuter wie chronischer Alkoholkonsum die Entladungsrate dopaminerger Neurone im dorsalen und ventralen Striatum [= Nucleus caudatus und Nucleus accumbens]. Bei protrahiertem Konsum [= langfristigem Konsum] wird die Adaption präsynaptischer Transport- und postsynaptischer Rezeptorensysteme induziert. Dabei können alkoholassoziierte Reize eine verstärkte präsynaptische Ausschüttung von Dopamin bewirken. Selbst kleine Mengen konsumierten Alkohols und Reize, die mit einem frühreren Alkoholkonsum verbunden sind, können somit eine verstärkte dopaminerge Neurotransmission auslösen. Das widerspiegelt sich als Alkoholverlangen oder verminderte Kontrolle über den Alkoholkonsum. So führte das Zeigen alkoholassoziierter Bilder zu einer neuronalen Aktivitätszunahme im meso-kortiko-limbischen System. Ein niedriges Ausmass der dopaminergen Produktion abstinenter alkoholabhängiger Patienten korrelierte mit Alkoholverlangen und der Trinkmenge im Rückfall. Ein Genotyp, der klinisch mit einem erhöhten Risiko einer schweren Entzugssymptomatik verbunden war, zeigte in vivo eine geringere Verfügbarkeit der Dopamintransporter im Putamen. Bei schnellen Änderungen der Dopaminfreisetzung (z. B. im Alkoholentzug) könnte eine verminderte Wiederaufnahmekapazität für Dopamin zu einer dopaminergen Überstimulation mit Entwicklung eines Delirs oder weiterer Entzugssymptome führen. Die Hypothese, dass Alkohol bei Individuen mit einem primär defizitären dopaminergen System zu protrahiertem Konsum führt, wird durch eine tierexperimentelle Untersuchung unterstützt. Eine Alkoholpräferenz bestand hier bei den Tieren mit erniedrigten Konzentrationen des Dopaminmetaboliten Homovanillinmandelsäure im Liquor und einer damit einhergehenden Hochregulation präsynaptischer Dopamintransporter im Striatum.“
In ihrer Disseration gibt die Psychologin Jennifer Uekermann eine Übersicht über die Ergebnisse verschiedener Untersuchungen zum Einfluss von Alkohol auf verschiedene Strukturen des Zentralnervensystems (Quelle: Jennifer Uekermann, Kognitive Veränderungen bei Alkoholismus und Depression, S. 22 ff. und 48 ff., Fakultät für Psychologie der Ruhr-Universität Bochum, 2002, http://www‑brs.ub.ruhr‑uni‑bochum.de/...).
Folgende neuropathologische Veränderungen struktureller und funktioneller Art sind demnach mit Hilfe moderner bildgebender Verfahren dokumentiert:
- Bei bis zu 70% der Alkoholiker findet man diffuse Veränderungen des Gehirns, z. B. eine Liquorvolumenerweiterung, die mit einem Zellverlust von Neuronen (graue Substanz) und Myelin-Gliazellen (weiße Substanz) einhergehen.
- Es zeigen sich bei Alkoholikern häufig generalisierte cortikale Hirnatrophien, die sowohl die Großhirnrinde als auch die Kleinhirnrinde betreffen. Von einer Atrophie des Kleinhirns ist ein großer Teil der Alkoholabhängigen betroffen, eine Kleinhirnataxie wurde bei ca. der Hälfte der Alkoholiker in einer Studie nachgewiesen.
- Bei Alkoholikern wurde im Vergleich mit einer Kontrollgruppe ein Neuronenverlust von 22% im oberen Frontalcortex festgestellt.
- Es wird über einen Neuronenverlust im frontalen Assoziationscortex und primären motorischen Cortex bei Alkoholikern berichtet.
- Bei Alkoholkranken findet man strukturelle Veränderungen der Basalganglien.
- Kommt es nicht zu einem Zelluntergang, ist eine Schrumpfung von Neuronen der Großhirnrinde eine weitere Möglichkeit der Schädigung durch Alkohol. Derartige Zellschrumpfungen werden für den oberen Frontalcortex und den Gyrus cinguli beschrieben.
- Unter Alkoholeinfluss kann es zu Dendritenveränderungen oder neuropathologischen Veränderungen im Parietalcortex, im Gyrus cinguli, im Hippocampus, am Corpus Callosum, den Basalganglien, der Amygdala und an den Mamillarkörpern kommen.
- Im Zusammenhang mit Alkoholeinfluss wurden Veränderungen der Empfindlichkeit des Hypothalamus-Hypophysen-Systems beobachtet.
- Im Zusammenhang mit Alkoholeinfluss wurde ein Verlust serotonerger Neurone der Raphekerne, die zur Struktur der Formatio reticularis von Medulla oblongata und Mittelhirn gehören, festgestellt.
- Experimentell wurde eine Reduktion cholinerger Neurone im basalen Vorderhirn und neuropathologische Veränderungen im Hippocampus, insbesondere im Gyrus dentatus, und im Kleinhirn bei unter Alkoholeinfluss stehenden Versuchstieren nachgewiesen.
Gefahren durch Alkohol betreffen Kinder, Jugendliche und junge Erwachsene bis zum 25. Lebensjahr in besonderem Maße, denn bis zu diesem Zeitpunkt befindet sich das Gehirn im Auf- und Umbau (→ Abschnitt 4.12.4, dort insbesondere Abbildung 40).
Es besteht unter anderem der Verdacht, dass die Myelinisierung der Neuronen während der Kindheit und Adoleszenz durch die zelltoxischen Eigenschaften des Alkohols stark behindert wird. Auch die direkten psychoaktiven Wirkungen des Alkohols können sich verheerend auf die jugendliche Hirnentwicklung auswirken. Insgesamt kann es keinen Zweifel daran geben, dass zumindest regelmäßiger Alkoholkonsum Jugendlicher die Gehirnentwicklung nachhaltig schädigt, ebenfalls Alkoholmissbrauch in Form des bei Jugendlichen beliebten „Komasaufens“. Neben anderen negativen Folgen ist damit ‑ zumindest aus kausaltheoretischer Sicht ‑ auch der späteren Entstehung einer Affektstörung Vorschub geleistet.
Vergleichbares gilt auch für die Schädigung der Leibesfrucht durch mütterlichen Alkoholkonsum während der Schwangerschaft. Dabei muss es nicht unbedingt zum Äußersten kommen, nämlich einer Erkrankung des Neugeborenen am Fetalen Alkohol-Syndrom (FAS) mit erheblichen Schädigungen seines Zentralnervensystems durch massive Alkoholeinwirkungen. Auch geringere Alkoholmengen können die embryonale Hirnentwicklung beeinträchtigen und die Gefahr einer Affektstörung im Verlaufe des späteren Lebens erhöhen.
Die Komorbidität zwischen Affektstörungen und Alkoholismus gilt emprisch als erwiesen. Die Ergebnisse verschiedener Komorbiditätsstudien weisen aufgrund sehr unterschiedlicher Studien-Designs allerdings eine große Spannbreite auf. So nennt J. Uekermann in ihrer Dissertation für Fragebogenstudien Quoten von 27 bis 69%. Sie verweist in ihrer Arbeit auch auf Studien, die bei Alkoholikern Defizite u. a. in der affektiven Reizverarbeitung feststellten, beispielsweise bei der Erkennung bestimmter affektiver sprachlich-tonaler Einfärbungen, auch affektive Prosodie genannt, was ebenfalls ein Indiz für einen Zusammenhang zwischen einer Alkohol- und Affekterkrankung ist (Quelle: Jennifer Uekermann, Kognitive Veränderungen bei Alkoholismus und Depression, wie oben).
Im Jahre 2004 erschien in einer Fachzeitschrift ein Beitrag von Michael Soyka, in dem Komorbiditätsraten von 24,3% bei Männern und 48,5% bei Frauen nach Prüfung der Ergebnisse verschiedener Studien als realistisch bezeichnet werden (Quelle: M. Soyka, M. Lieb, Depression und Alkoholabhängigkeit - Neue Befunde zur Komorbidität, Neurobiologie und Genetik, Journal für Neurologie, Neurochirurgie und Psychiatrie, 5 (3), 2004, Krause & Pachernegg GmbH, Verlag für Medizin und Wirtschaft, Gablitz/Österreich 2004, http://www.kup.at/...).
Die über Suchtmechanismen und Zusammenhänge zwischen einem Suchtstoff und psychiatrischen Erkrankungen zu Beginn des Abschnitts 4.13.8 gemachten Modellaussagen gelten ohne Einschränkungen auch für den Alkohol.
Die Komorbidität alleine ‑ sei sie auch noch so hoch ‑ ist kein Beweis für Ursache-Wirkungs-Beziehungen. Zur Diskussion der speziellen Ursachen der Komorbidität zwischen Alkoholsucht und Affektstörungen, bei der neben biologischen Faktoren auch soziale Umstände, die Umgebung oder psychologische Einflüsse ein Rolle spielen können, haben sich folgende vier Modelle etabliert (Quelle: Jennifer Uekermann, Kognitive Veränderungen bei Alkoholismus und Depression, S. 18, wie oben):
- Zufallsmodell, bei dem Alkoholismus und Affektstörung unabhängig voneinander auftreten.
- Ätiopathogenese-Modell mit den drei Varianten (1) Alkoholismus ist die Ursache einer anderen ‑ dritten ‑ Störung, aus der wiederum die Affektstörung resultiert, (2) Affektstörung als Ursache für die Entstehung des Alkoholismus und (3) Alkoholismus als direkte Ursache der Affektstörung.
- Phänomenologisches Modell mit der Annahme, dass eine gemeinsame Ursache („hidden variable“) für beide Erkrankungen angenommen wird, beispielsweise polygenetische Zusammenhänge.
- Interaktionales Modell mit der Annahme, dass eine Affektstörung den Verlauf einer Alkoholabhängigkeit direkt oder indirekt beeinflusst.
Alle vier Modelle lassen sich nach dem Muster des allgemeinen Teufelskreismodells (→ Abbildung 46) integrieren. Das weist darauf hin, dass jede Sichtweise ihre Berechtigung hat.
Das Zufallsmodell ist im Rahmen des Teufelskreismodells jedoch nur als eine kurzfristige Erklärung für den Einstieg in die Sucht zu rechtfertigen, denn mittel- bis langfristig treten Abhängigkeiten und Interaktionen zwischen beiden Erkrankungen auf. Eine rein von Zufällen ausgehende Sichtweise scheidet daher bei einer längerfristigen Betrachtung aufgrund dieser Wechselwirkungen als Erklärungsmuster aus.
Über diese vier Modelle hinaus ist noch die Möglichkeit einer Affekstörung nach Suchtstoffentzug zu berücksichtigen; diese Variante wird vom Teufelskreismodell nicht abgebildet.
Im nun folgenden letzten Teil dieses Abschnitts soll es um die Frage gehen, ob Alkohol eine Affektstörung mitverusachen kann, da die Komorbiditätsrate alleine für eine Aussage über Ursache‑Wirkungs‑Beziehungen bekanntlichermaßen ungeeignet ist. Ein solches Szenario entspricht - je nach Sichtweise - den Varianten 1 oder 3 des oben beschriebenen Ätiopathogenese-Modells:
Ätiopathogenese-Modell, Variante 1: Alkoholkrankheit → Drittstörung → Affektstörung
Ätiopathogenese-Modell, Variante 3: Alkoholkrankheit → Affektstörung
Zwei verschiedene Begründungen kommen dafür infrage. Die erste Begründung ist die Annahme, ein Zusammenhang sei rein psychologisch ohne jegliche neurobiologische Komponente begründet. Anhänger dieser These führen als Beispiel die Sorge des Betroffenen über seine Alkoholkrankheit an, die in der Folge dann in einer Affektstörung mündet. Diese Sichtweise entspricht am ehesten der Variante 3 des Ätiopathogenese-Modells.
Diese Sichtweise kann aus kausaltheoretischer Sicht nicht zutreffen, denn eine Affektstörung hat nach dieser immer einen neurobiologischen Hintergrund. Einzig eine depressive Verstimmung kann psychologisch begründet werden. Eine depressive Verstimmung darf jedoch nicht mit einer Affektstörung verwechselt werden. Beide affektiven Zustände sind streng voneinander zu trennen.
Die zweite ‑ kausaltheoretisch fundierte ‑ Begründung unterstellt einen Ursache-Wirkungs-Zusammenhang aufgrund neurobiologischer Veränderungen bzw. Schäden und kommt damit Variante 1 des Ätiopathogenese-Modells am nächsten, bei der neurologische Alkoholfolgen dann als Drittstörung gelten:
Alkoholkrankheit → Neurobiologische Veränderungen/Schäden → Affektstörung
Die Begündung dafür ist naheliegend, denn viele der in Abbildung 3 als affektrelevant bezeichneten Areale (→ Abbildung 3 in Kapitel 1) spielen beim Alkoholismus eine Rolle und darüber hinaus sind Zelltoxizität und Psychoaktivität des Alkohols bekannt. Die oben aufgelisteten neuropathologischen Befunde bei Alkoholismus haben mit den als Schlüsselregionen einer Affektstörung verdächtigten Bereichen (→ Abschnitt 1.3) bzw. durch diagnostische Verfahren identifizierten Bereichen (→ Abschnitt 1.6) eine maximale Schnittmenge. Es existiert damit keine affektrelevante Hirnregion, die nicht durch chronischen Alkoholkonsum gefährdet ist.
Es bleibt noch zu prüfen, ob es für die kausaltheoretische Sichtweise von Alkohol und Affektstörungen auch empirische Belege gibt und ob solche Belege Genaueres über die Zusammenhänge zwischen Alkohol und Affektstörungen preisgeben, beispielsweise eines neurologischen Zusammenhangs.
Interessant sind zunächst Ergebnisse aus dem hier schon zitierten Artikel von Michael Soyka und M. Lieb über unterschiedliche Prävalenzraten bei primären und sekundären depressiven Syndromen (Quelle: M. Soyka, M. Lieb, Depression und Alkoholabhängigkeit - Neue Befunde zur Komorbidität, Neurobiologie und Genetik, Journal für Neurologie, Neurochirurgie und Psychiatrie, 5 (3), 2004, Krause & Pachernegg GmbH, Verlag für Medizin und Wirtschaft, Gablitz/Österreich 2004, http://www.kup.at/...). Danach ergab sich bei verschiedenen klinischen Studien eine „Häufigkeit depressiver Symptome bei Alkoholikern mit 3 bis 98% (...), wobei die Prävalenzraten meist zwischen 30 und 60% lagen. Für primäre depressive Symptome wurden bei Alkoholkranken Prävalenzraten zwischen 2 und 12% ermittelt, während sekundär ‑ zur bestehender Alkoholabhängigkeit hinzutretende ‑ depressive Syndrome eine Prävalenz von 12 bis 51% hatten.“ Die stark unterschiedlichen Ergebnisse resultierten aus unterschiedlichen Rahmenbedingungen und voneinander abweichenden Studien-Designs. Teilweise ergaben sich Prävalenzraten von bis zu 74% für eine sekundäre Depression, wobei hier Männer häufiger betroffen waren. Dennoch lässt sich eine Tendenz für häufiger auftretende sekundäre depressive Syndrome im Vergleich mit primären depressiven Syndromen erkennen. Das bedeutet: Auf eine schon bestehende Alkoholabhängigkeit folgt demnach häufiger eine Affekterkrankung als umgekehrt Alkoholabhängigkeit einer schon bestehenden Affektstörung folgt.
Letzteres ist zwar kein Beweis für eine Kausalität zwischen Alkohol als auslösendem Faktor und einer Affektstörung, jedoch ein starkes Indiz dafür.
Indizien eines Kausalzusammenhangs zwischen Alkohol und dem Auftreten einer Depression liefert die empirische Ermittlung von Prävalenzraten in Verbindung mit Alkoholentzugs- und Alkoholentwöhnungstherapien:
- Insgesamt weisen sechs Quellen (Nr. 23 bis 28 in der Literaturangabe von Soyka und Lieb) auf höhere Prävalenzraten in Phasen hohen Alkoholkonsums und während Entgiftungsbehandlungen hin bzw. demgegenüber auf niedrigere Prävalenzraten während rehabilitatorischer Entwöhnungsmaßnahmen (diese Patienten haben sich schon einer erfolgreichen Alkoholentzugstherpaie unterzogen) und bei längerer Abstinenz.
- In eine ähnliche Richtung deuten Ergebnisse verschiedener Vorher-Nacher-Untersuchungen. In einer Studie wiesen 62% der Patienten vor einer Alkoholentzugstherapie eine Major Depression auf, während es danach nur noch 13% waren (Quelle Nr. 29). Verlaufsuntersuchungen kamen zu vergleichbaren Resultaten. Die Skalenwerte der Depressionsdiagnose besserten sich im Verlauf einer Entzugsbehandlung signifikant und bei Entwöhnungsbehandlungen wurde fast immer eine Abnahme der Skalenwerte verzeichnet. In einer Quelle traten nur bei rückfälligen Alkoholikern vermehrt depressive Syndrome auf (Quellen Nr. 30 bis 32).
- Die Gefahr der Fehlinterpretation von Untersuchungergebnissen im Zusammenhang mit Entzugs- oder Rehabilitationsmaßnahmen verneinen die US-amerikanischen Epidemiologinnen Deborah Hasin und Bridget F. Grant in ihrer Arbeit mit 6.050 trockenen Alkoholikern (Quelle Nr. 34, D. S. Hasin, B. F. Grant, Major Depression in 6050 Former Drinkers, JAMA Psychiatry/Archives Of General Psychiatry, 59, 9/2002, American Medical Association, Chicago, Illinois, USA, http://www.columbia.edu/...). Eine Depressivität von Studienteilnehmern könnte nämlich auch durch Entzugs- oder Vergiftungssymptome begründet sein und nicht aufgrund Alkohols. Das Ergebnis war eindeutig: „Sie untersuchten eine Stichprobe von Alkoholkonsumenten (...) . Es wurden 6.050 ehemalige Alkoholkonsumenten, die innerhalb des letzten Jahres wenig bzw. gar nicht getrunken, nicht geraucht und keine psychotropen Substanzen konsumiert hatten, auf den Zusammenhang zwischen Alkoholabhängigkeit und späterer depressiver Störung untersucht. Frühere Alkoholabhängigkeit erhöhte danach das Risiko für Episoden einer Major Depression (DSM-IV) um den Faktor 4.“
Weiter zitieren Soyka und Lieb auch aus Arbeiten, bei denen es um neurobiologische Gemeinsamkeiten von Alkoholismus und depressiven Störungen geht. Sie erwähnen eine besonders ausführliche Zusammenfassung wichtiger Studien mit 98 Quellenverweisen durch José J. Miguel‑Hidalgo und Grazyna Rajkowska, Mitarbeiter der Psychiatrischen Abteilung des Universitätskrankenhauses der University of Mississippi (Quelle Nr. 36, J. J. Miguel‑Hidalgo, G. Rajkowska, Comparison of prefrontal cell pathology between depression and alcohol dependence, Journal of Psychiatric Research, 2003, 37 (5), S. 411 - 420, Elsevier B.V., Amsterdam 2003, https://www.ncbi.nlm.nih.gov/...). Miguel-Hidalgo und Rajkowska weisen in ihrem Aufsatz auf übereinstimmende Befunde verschiedener Untersuchungen hin, bei denen sich Läsionen im präfrontalen Cortex ergaben. Gliazellen scheinen sowohl bei Alkoholismus in Verbindung mit einer Depression als auch bei einer alleinigen Depression wesentlich stärker betroffen zu sein als Nervenzellen. Dennoch sind strukturelle gegensinnige Unterschiede erkennbar. Während beim Alkoholismus Gliazellkerne schrumpfen ‑ vermutlich durch zytotoxische Effekte des Alkohols ‑, sind diese bei einer Depression ohne Alkoholismus vergrößert, wobei hier keine Ursachen für die Größenveränderung feststellt werden konnten. Es stellte sich aber heraus, dass die Dysfunktionalität der Gliazellen in beiden Fällen die Depression verursachen.
Auch diese Ergebnisse harmonieren mit wichtigen kausaltheoretischen Postulaten. Erstens ist das ein deutlicher Hinweis für die Beteiligung pathologisch veränderter Gliazellen an der Genese von Affektstörungen, und zweitens spielt es demnach keine Rolle, auf welchen pathologischen Mechanismen diese Veränderungen genau beruhen. Entscheidend ist nur, ob ein bestimmtes Hirnareal seine Informationsverarbeitungsfunktionen aufgrund von Zellprozessstörungen nicht mehr bewältigen kann.
Alle anderen von Miguel-Hidalgo und Rajkowska untersuchten Studien lassen es jedoch nicht zu, genaue Ursache-Wirkungs-Zusammenhänge abzuleiten, sie bestätigen lediglich schon die bekannte Komorbidität.
Die Suche nach gemeinsamen polygenetischen Ursachen brachte ebenfalls bisher keine eindeutigen Ergebnisse. Die beiden Autoren äußern sich darüber hinaus skeptisch über die Möglichkeit, jemals einen genetischen Marker für Alkoholismus sicher etablieren zu können. Diese Feststellung deckt sich mit Ergebnissen genetischer Analysen bei Affektstörungen (→ Abschnitt 4.7.6).
Welches Fazit ergibt sich aus den theoretischen, neuropathologischen und empirischen Erkenntnissen über das Verhältnis zwischen Alkoholismus und Affektstörungen? Kann Alkohol als eine neurobiologische Noxe definiert werden, die affektive Störungen mitverantwortet?
Die wissenschaftliche Forschung zeigt klar, dass Alkohol ‑ insbesondere bei längerfristigem Konsum ‑ das Zentralnervensystem schädigt. All dies legt eine Mitverantwortung des Alkohols an der Genese auch von Affekterkrankungen nahe:
- Alkohol beeinflusst Transmissionssysteme nachhaltig negativ, die in der Affektverarbeitung eine Rolle spielen.
- Alkohol führt nachhaltig zu funktionalen und strukturellen Veränderungen in sämtlichen affektrelevanten Hirnarealen.
- Die besondere Gefahr des Alkohols für Jugendliche ist mit der langen Entwicklungszeit des Gehirns bis zum 25. Lebensjahr zu begründen, die durch Alkohol nachhaltig gestört wird. Vergleichbares gilt auch für den mütterlichen Alkoholkonsum während der Schwangerschaft für die Embryonalentwicklung des Nervensystems.
Auch empirische Untersuchungen weisen auf die Existenz eines Kausalzusammenhangs zwischen Alkohol bzw. Alkoholismus als einem Mitauslöser für Affektstörungen hin, wenngleich die Beweise dafür noch nicht endgültig erbracht werden konnten. Stellt man folgende Studienergebnisse jedoch den genannten neuropathologischen Befunden gegenüber, lässt sich ein derartiger Zusammenhang kaum noch abstreiten:
- Empirisch wurde eine hohe Komorbidität zwischen langfristiger Alkoholabhängigkeit und Affektstörungen festgestellt.
- Die Wahrscheinlichkeit, an einer Affektstörung zu erkranken, ist für Alkoholiker wesentlich höher als die Wahrscheinlichkeit für affektiv Erkrankte, Alkoholiker zu werden (Prävalenz).
- Pathologische Gliazellveränderungen in affektrelevanten Hirnarealen lösen mit hoher Wahrscheinlichkeit Affektstörungen aus.
Der Nachweis, dass sowohl die Neigung zum Alkoholismus als auch die Neigung zur affektiven Erkrankung auf gemeinsamen polygenetischen Ursachen fußen, somit korrelativ polygenetisch miteinander in Beziehung stehen, konnte weder erbracht werden noch scheint es überhaupt jemals möglich zu sein, einen solchen Zusammenhang belegen zu können (→ vgl. Abschnitt 4.7.6).
Tetrahydrocannabinol (THC)/Cannabis
Delta-8- bzw. insbesondere Delta-9-Tetrahydrocannabinol ‑ kurz THC ‑ sind die mit Abstand stärksten psychoaktiven Substanzen bestimmter Sorten der weiblichen Hanfpflanze Cannabis und werden als Cannabinoide bezeichnet. Dazu gehören unter anderem auch Cannabigerol (CBG) und Cannabidiol (CBD), jedoch reicht keine von ihnen an die Wirkungsstärke und das Wirkungsprofil von THC heran. THC wird als Inhaltsstoff des gepressten Cannabisharzes (Hasch, Haschisch) oder getrockneter Cannabisblüten (Marihuana, „Gras“) als Rauschmittel konsumiert.
Der THC-Gehalt in Cannabispflanzen schwankt stark und hängt von der Aufzuchtmethode ab. So entstehen aus Kreuzungen verschiedener Cannabissorten Pflanzen mit hohem THC-Gehalt, die als „Skunk-Cannabis“ bezeichnet werden. Herstellung, Verkauf und Besitz von Cannabis ist in den meisten Ländern verboten.
Während der Grund für die Nikotinproduktion der Tabakpflanze geklärt ist (→ Abschnitt Nikotin/Tabakrauchen), gibt es über den Sinn der pflanzlichen THC-Synthese verschiedene Theorien, von denen noch keine bewiesen wurde. So könnte THC ein natürlicher Pflanzenschutz gegen Krankheiten, Fressfeinde oder die schädlichen Folgen übermäßiger Sonneneinstrahlung sein. Auch wenn sich dieser Gedanke aufdrängt: Die psychoaktive Wirkung von THC steht mit seiner pflanzlichen Produktion in keinem Zusammenhang, denn es verwandelt sich erst durch Verbrennen bzw. Erhitzen in ein Rauschmittel.
THC wird daher meist zusammen mit Tabak als „Joint“ oder mit einer Wasserpfeife rauchend inhaliert, obwohl Nikotin dessen Wirkung abschwächt. Eine Wasserpfeife („Bong“) steigert die Menge des auf einmal zu inhalierenden THCs erheblich. Bei der Aufnahme über die Lunge setzt die Wirkung schon nach wenigen Minuten bis zu einer Viertelstunde ein.
Alternativ erfolgt die Aufnahme in den Körper durch Resorption im Verdauungstrakt, meist mit THC-haltigen Teegetränken („Marihuana-Tee“) oder Mehlspeisen („Space Cookies“), denn auch so ist die Voraussetzung des Erhitzens durch Kochen oder Backen erfüllt. Die Wirkungszeit ist hier im Vergleich mit der Aufnahme über die Lunge wesentlich länger, die „Wartezeit“ beträgt im Schnitt zwei Stunden, was jedoch auch von der Nahrungsverdauung abhängt. Auf nüchternen Magen setzt die Wirkung schneller ein, bei gefülltem Magen dauert es entsprechend länger.
Die Wirkungsdauer beträgt beim Inhalieren etwa zwischen einer und drei Stunden und bei der oralen Aufnahme sechs Stunden und mehr ‑ je nach THC-Menge.
Darüber hinaus hängen Wirkungszeit und Wirkungsdauer von weiteren Kriterien ab, beispielsweise Konsumhäufigkeit, Situation und persönlicher Disposition.
Die Art und Weise bzw. Qualität der Rauschwirkung steht ebenfalls mit allen genannten Kriterien in einem Zusammenhang. Bei den kurzfristigen Rauschwirkungen handelt sich in erster Linie um Veränderungen des Bewusstseins und der Wahrnehmung, einschließlich Veränderungen des affektiven Erlebens. Kurzfristig bedeutet, dass die Symptome mit Nachlassen der pharmakologischen THC-Wirkung abklingen.
Die Wirkungen sind von Person zu Person unterschiedlich bzw. werden unterschiedlich wahrgenommen und führen zu einer erheblichen Beeinträchtigung der Leistungsfähigkeit. Es handelt sich zum Beispiel um folgende Veränderungen:
- Verstärkung des assoziativen Denkens
- Gefühl, zu allumfassenden und tiefen Erkenntnissen zu gelangen
- Veränderungen der Wahrnehmung von Dingen bzw. der eigenen und anderen Personen
- Gedanken werden sprunghaft, um dann wieder hartnäckig um ein Thema zu kreisen
- Veränderte Zeitwahrnehmung, so dass Stunden zu Sekunden werden können und umgekehrt
- Störungen des Kurzzeitgedächtnisses
- Aufmerksamkeitsstörungen
- Positive Gefühle können verstärkt werden, bis hin zu Euphorie und Enthemmung
- Negative Gefühle können verstärkt werden, bis hin zur Angstattacke und/oder Depression
- Affektive Verflachung
- Intensivierung der Sinneswahrnehmungen
- Halluzinationen/Trugwahrnehmungen
- Misstrauen oder paranoide Wahnvorstellungen*
* Diese Symptome können ggf. auch bis zu einer Woche andauern (Quelle: E. Hoch, R. Thomasius, F. Ganzer et al., Risiken bei nichtmedizinischem Gebrauch von Cannabis, Deutsches Ärzteblatt, 112 (16), erschienen am 17.4.2015, Deutscher Ärzteverlag GmbH, Berlin 2015, https://www.aerzteblatt.de/...).
Außerdem sind noch weitere kurzfristige psychogenen Reaktionen möglich:
- Gesteigerter Redefluss bzw. Logorrhoe
- Anlasslose Lachanfälle
- Neigung zur Albernheit
- Entspannung, Antriebsverlust und Motivationsstörungen
- in seltenen Fällen Agitiertheit
- Konzentrations- und Lernstörungen
- Beeinträchtigung der Reaktionszeit
- Orientierungslosigkeit
- Gesteigertes Nahrungsaufnahmebedürfnis
THC soll darüber hinaus auch eine schmerz- und krampflösende Wirkung haben, so dass im Zusammenhang mit medizinischen Cannabis-Anwendungen eine Kombination von THC und einem anderen Cannabinoid des Hanfs ‑ Cannabidiol (CBD) ‑ diskutiert wird, denn CBD wirkt hauptsächlich schmerz- und krampflösend.
Die grundsätzlichen neurobiologischen Wirkungsmechanismen von THC ähneln denen anderer psychotroper Substanzen: Körperfremde Substanzen verdrängen körpereigene Substanzen und stören dadurch die Reizverarbeitung im Gehirn. Das körperfremde THC bindet vor allem an den beiden Cannabinoid-Rezeptoren CB1 und CB2, die für die körpereigenen Endocannabinoide Anandamid und 2‑Arachidonylglycerol vorgesehen sind, verdrängt diese und stört damit die natürliche und sehr empfindliche Nervenreizübertragung. THC wird als Partialagonist bezeichnet, da es die CB1-Signalübertragungsmechanismen nach der „feindlichen“ Besetzung des Rezeptors nur unvollständig auslöst. THC wirkt allerdings ‑ abhängig von der Dosis ‑ meist wesentlich stärker. Neben Anandamid und 2‑Arachidonylglycerol gibt es noch das körpereigene Virodhamin, das jedoch nur antagonistisch wirkt, das heißt die beiden anderen Endocannabinoide in ihrer Wirkung lediglich hemmt, indem es die Rezeptoren besetzt und keine eigene Wirkung entfaltet.
In Gehirn und Rückenmark sind die CB1‑Rezeptoren lokalisiert. An den psychogenen Wirkungen sind daher naturgemäß nur sie beteiligt, während man dagegen CB2‑Rezeptoren außerhalb des Zentralnervensystems in Immun- oder Knochenmarkzellen, Milz, Thymus oder im Darm findet.
CB1-Rezeptoren befinden sich überwiegend an den präsynaptischen Axonendungen von Neuronen im Gehirn und müssen daher dort ihren Haupteinfluss haben. Der natürliche CB1-Grundmechanismus besteht darin, die Freisetzung anderer Botenstoffe an genau diesen präsynaptischen Axonendungen zu hemmen. Die natürlichen Endoannabinoide, die in der postsynaptischen Nervenzelle synthetisiert werden, wirken damit über den synaptischen Spalt auf die präsynaptischen CB1-Rezeptoren zurück. Da die Neurotransmitterübertragung normalerweise von der präsynaptischen auf die postsynaptische Nervenzelle erfolgt, wird die umgekehrte Form als „retrograde synaptische Übertragung“ bezeichnet, der englische Fachbegriff lautet „Retrograde signaling“.
Da die körpereigenen Endocannabinoide nicht völlig selektiv sind, beeinflussen sie neben CB1 und CB2 in zweiter Linie noch eine Reihe anderer Rezeptoren, dort jedoch in wesentlich geringerem Umfang und wahrscheinlich auf der Basis eines anderen Mechanismus. Ebenfalls wird die Existenz weiterer Cannabinoid-Rezeptoren vermutet, die in einer ähnlichen Art und Weise Cannabinoide für die Reizverarbeitung nutzen wie CB1 und CB2.
Aufgrund des ubiquitär-heterogenen Vorkommens der CB1-Rezeptoren in allen Hirnarealen ‑ in der Medulla oblongata, im Mittelhirn, in Thalamus und Hypothalamus, in den Basalganglien, in der Amygdala, im Hippocampus, im Cerebellum (Kleinhirn) und auch in fast allen Bereichen des Cortex ‑ und der nicht völlig selektiven Bindungsfreudigkeit der Endocannabinoide lassen sich die vielen Auswirkungen körpereigener Cannabinoide erklären. Einige dieser Regionen sind besonders gut mit CB1 versorgt, „wobei insbesondere Cerebellum, Teile der Basalganglien, Hippokampus und viele Regionen des Neokortex eine besonders hohe Dichte an CB1 Rezeptoren aufweisen (...).“ (Quelle: Benjamin Becker, Die Effekte von Cannabis und Ecstasy auf die neurokognitive Hirnfunktion beim Menschen, Inaugural-Dissertation, Institut für Experimentelle Psychologie der Heinrich-Heine-Universität Düsseldorf, Düsseldorf 2010, http://docserv.uni-duesseldorf.de/...).
Da sich also CB1-Rezeptoren in vielen Regionen des Gehirns befinden, betrifft THC entsprechend viele Neurotransmittersysteme. CB1-Rezeptoren beeinflussen Neurone, die zum Beispiel GABA, Glutamat, Acetylcholin oder Noradrenalin ausschütten, und eine CB1-Aktivierung durch THC hemmt dann die Freisetzung dieser Botenstoffe. Carsten Wotjak, Mitarbeiter am Münchener Max-Planck-Institut für Psychiatrie, beschreibt die Wirkungen des endocannabinoiden Transmittersystems daher treffend als „bunt wie ein Wiesenblumenstrauß“, was genau an dieser weiten Verbreitung von CB1-Rezeptoren im Gehirn und deren Involvierung in fast alle Transmittersysteme liegt (Quelle: Ulrich Pontes, Endocannabinoide - Kommunikation im Gehirn, Neurowissenschaftliche Gesellschaft e. V., Berlin, 2012, https://www.dasgehirn.info/...). Und genau deshalb sind die Wirkungen des exogenen THCs ebenfalls entsprechend differenziert und „bunt wie ein Wiesenblumenstrauß“, was auch der Aufzählung der vielen unterschiedlichen Cannabisauswirkungen zu Beginn des Abschnitts zu entnehmen ist.
Die Hirnareale mit besonders vielen CB1-Rezeptoren werden mit folgenden kognitiv-emotionalen und motorischen Funktionen in Verbindung gebracht, die damit alle durch THC beeinflusst werden:
- Hippocampus → Gedächtnisfunktionen, Lernprozesse
- Basalganglien → Grobmotorik, Belohnungssystem
- Kleinhirn → Feinmotorik
- Mittelhirn, Thalamus, Hypothalamus, Basalganglien, Amygdala, Hippocampus, untere Cortexbereiche, obere Cortexbereiche/Assoziationsareal → Emotionsverarbeitung
- Amygdala → Angstverarbeitung
- Mittelhirn → Antrieb
- Obere Cortexbereiche → Bewusste Informationsverarbeitung
- Neuronale Netze, z. B. die Formatio reticularis → Generelle Informationsverarbeitung
Dass THC Konsumenten kurzfristige affektive Probleme bis hin zu einer Depression bescheren kann, ist vielen Konsumentenberichten zu entnehmen und gängige Lehrmeinung. Diese Erfahrungen stehen auch mit den hier vertretenen Standpunkten im Einklang, beispielsweise kurzfristigen Auswirkungen THC-bedingter funktionaler Störungen in affektrelevanten Hirnarealen, die im Modell mit der ersten Stufe des 3‑Stufen‑Modells begründet werden können (→ Abschnitt 1.4). Kurzfristige depressive Stimmungen oder Störungen durch THC treten jedoch erfahrungsgemäß vor allem durch Verstärkung einer vorhandenen depressiven Grundstimmung auf, die kurz vor dem THC-Konsum schon bestand.
Bei der Diskussion um THC stehen aber vor allem die langfristige Wirkungen im Mittelpunkt, drehen sich Fragen hier doch vor allem um eventuelle hirnorganische Schäden durch wiederholten THC-Konsum und/oder durch den THC-Konsum Jugendlicher während der sensiblen Phase ihrer Hirnentwicklung, aufgrund dessen sich später eine Suchterkrankung oder andere psychiatrisch-neurologische Erkrankungen oder Auffälligkeiten, insbesondere Affektstörungen, manifestieren könnten.
Für eine Einschätzung dieser Zusammenhänge sind ‑ neben kausaltheoretischen Begründungen ‑ auch Ergebnisse empririscher Untersuchungen hilfreich.
Bei der Untersuchung langfristiger Wirkungen von THC auf Gehirn und Psyche treten leider die gleichen Probleme auf, die auch von den Untersuchungen anderer psychogener Substanzen bekannt sind, und auf die zu Beginn des Abschnitts mit der Darstellung des Teufelskreismodells schon hingewiesen wurde (→ Abbildung 46): Hat eine schon bestehende psychiatrische Erkrankung oder die Neigung zu depressiven Verstimmungen zum Cannabiskonsum geführt oder macht „Kiffen“ depressiv oder haben beide Erklärungsalternativen im Sinne eines „Teufelskreises“ Relevanz?
Die Cannabisabhängigkeit ist als Erkrankung offiziell anerkannt. Auf der Liste der psychischen Erkrankungen und Verhaltensstörungen nach der internationalen ICD‑10/GM 2017 hat der Cannabiskonsum unter dem Rauschwirkungsaspekt drei Notationen: F12.1 als Cannabismissbrauch, F12.2 als Cannabisabhängigkeit und F12.7 für langandauernde Störungen durch Cannabis. Darüber hinaus gibt es noch die Notation T40.7 für somatische Vergiftungserscheinungen von Cannabis oder Cannabisderivaten. Auch die US-amerikanische Alternative zur ICD‑10, die DSM‑5, klassifiziert die cannabisbedingten Konsumstörungen Abhängigkeit und Missbrauch und substanzeninduzierte Störungen bei Cannabiskonsum mit den Ausprägungen Intoxikation, Entzug und Delir. Eine erhebliche Störung bzw. ein problematisches Konsummuster wird gemäß DSM‑5 diagnostiziert, wenn mehr als drei von elf zur Auswahl stehenden Symptombeschreibungen zutreffen. Einige davon betreffen Umstände, die direkt mit einer Abhängigkeit assoziiert werden, zum Beispiel den Konsum größerer Mengen als beabsichtigt, erfolglose Entzugs- oder Reduktionsversuche oder Konsum trotz dadurch verursachter sozialer und familiärer Probleme.
Eine direkte empirische Untersuchung langfristiger THC-Wirkungen auf Menschen mit einer prospektiv-experimentellen Studie wäre die Lösung, ist aus ethischen Gründen jedoch völlig zu Recht ausgeschlossen. Außerdem müsste eine solche Studie, um wirklich eindeutige Rückschlüsse zuzulassen, als Interventionsstudie angelegt sein, also unter weitgehender Kontrolle aller Einflussfaktoren stattfinden, was aber praktisch sehr schwierig ist. Tierexperimente sind ebenfalls ethisch fragwürdig, und deren Ergebnisse auch nicht mit völliger Sicherheit auf Menschen übertragbar. Kompromisse sind hier ‑ wie immer ‑ möglichst gut strukturierte prospektive Beobachtungsstudien, eher ungünstiger sind retrospektive Studien aufgrund ihrer meist unsicheren Datengrundlage.
Trotz aller Schwierigkeiten wird intensiv geforscht, inwiefern Cannabiskonsum Auswirkungen auf das Gehirn hat, sowohl auf dessen kognitive und affektive Leistungen, als auch bezüglich hirnorganisch-funktionaler bzw. langfristig hirnorganisch-struktureller Veränderungen generell. Um es gleich vorwegzunehmen: Die Ergebnisse sind jedoch häufig uneinheitlich.
Benjamin Becker vergleicht in seiner schon oben zitierten Dissertation aus dem Jahre 2010 verschiedene Studienergebnisse: „Eine steigende Anzahl wissenschaftlicher Publikationen berichtet von kognitiven Defiziten bei Cannabis-Konsumenten (Grant et al., 2003, Gonzalez, 2007, Solowij and Battisti, 2008). Im Vergleich zu Kontrollen zeigten chronische Konsumenten Beeinträchtigungen in den Bereichen episodisches Gedächtnis, Arbeitsgedächtnis, Aufmerksamkeit und exekutive Funktionen (Block and Ghoneim, 1993, Bolla et al., 2002, Solowij et al., 2002a, Solowij et al., 2002b, Messinis et al., 2006). Da die Hauptwirksubstanz des Cannabis, THC, noch Tage bis Wochen nach dem letzten Konsum im Körper von chronischen Konsumenten nachweisbar ist (McGilveray, 2005), bleibt allerdings unklar, ob es bei den berichteten Defiziten um subakute und somit potentiell reversible oder um länger andauernde Folgewirkungen des Cannabis-Konsums handelt. Die Befundlage hierzu ist uneinheitlich, so berichten Studien mit einer Abstinenzzeit von bis zu 28 Tagen teils von einer Normalisierung der Leistungsfähigkeit (Schaeffer et al., 1981, Pope et al., 2001a), teils von andauernden Defiziten (Bolla et al., 2002, Solowij et al., 2002b). Eine Metaanalyse von Grant (Grant et al., 2003) und ein aktuelles Review von Solowij und Battisti (Solowij and Battisti, 2008) finden die konsistentesten Befunde für Langzeiteffekte vor allem im Bereich Gedächtnis. Die kognitiven Defizite konnten in mehreren Studien mit spezifischen Parametern des Cannabis-Konsums assoziiert werden. Zusammenhänge zwischen einer schlechteren kognitiven Leistung zeigten sich dabei v. a. mit einem früheren Einstiegsalter (Ehrenreich et al., 1999, Pope et al., 2003), einer längeren Dauer des Konsums (Solowij et al., 2002a, Messinis et al., 2006) und einer höheren Konsumhäufigkeit (Pope and Yurgelun-Todd, 1996, Pope et al., 2001b). In den letzten Jahren wurden verstärkt Untersuchungen mittels funktioneller Magnetresonanztomographie durchgeführt, um die neuronalen Grundlagen der kognitiven Auffälligkeiten bei Cannabis-Konsumenten zu untersuchen (Quickfall and Crockford, 2006, Chang and Chronicle, 2007, Martin-Santos et al., 2010). Im Vergleich zu Kontrollen zeigten abstinente chronische Konsumenten trotz unauffälliger Leistung veränderte Hirnaktivierung bei einer Reihe kognitiver Funktionen, wie etwa assoziatives Lernen (Jager et al., 2007b, Nestor et al., 2008), Aufmerksamkeit (Chang et al., 2006) und Arbeitsgedächtnis (Kanayama et al., 2004, Jager et al., 2006). Zumeist wurden stärkere neuronale Aktivierung innerhalb der beteiligten neuronalen Netzwerke und/oder nicht primär aufgabenrelevanten Hirnregionen bei den Konsumenten von Cannabis berichtet. Im aktuellsten Review über die Auswirkungen von Cannabis auf Hirnaktivierung interpretieren Martin-Santos und Mitarbeiter (Martin-Santos et al., 2010) diese Hyperaktivität bei unauffälliger kognitiver Leistung bei Cannabis-Konsumenten als kompensatorischen Mechanismus im Sinne der 'cognitive efficiency hypothesis' (Vernon, 1983, Rypma and D'Esposito, 2000). Demnach benötigen die Cannabis-Konsumenten höhere neurokognitive Ressourcen, um eine Leistungsfähigkeit im normalen Bereich aufrecht zu erhalten.“ (Quelle: Benjamin Becker, Die Effekte von Cannabis und Ecstasy auf die neurokognitive Hirnfunktion beim Menschen, Inaugural-Dissertation, Institut für Experimentelle Psychologie der Heinrich-Heine-Universität Düsseldorf, Düsseldorf 2010, http://docserv.uni‑duesseldorf.de/...).
Die von Becker zitierten Studien der Jahre 1981 bis 2010 unterlagen naturgemäß keiner einheitlichen Methodik. Bei einer Untersuchung chronischer Cannabis-Konsumenten ist davon auszugehen, dass die Einnahme weiterer psychotroper Substanzen (Nikotin, Alkohol, andere Rauschmittel) einen erheblichen Störfaktor für die Auswertungen darstellt. Die zum Schluss genannten relativ neueren Studien der Jahre 2004 bis 2010, bei denen mit einer funktionellen Magnetresonanztomographie (fMRT) THC-bedingte Auswirkungen auf die neuronale Hirnaktivierung gemessen wurden, erlauben nach Aussage von Becker jedoch „keine gesicherte Aussage über mögliche langfristige Auswirkungen des Cannabis-Konsums“.
Aus diesen Gründen führte Becker an der Universität zu Köln zwei fMRT-Studien durch, die eindeutigere Aussagen über die langfristigen hirnorganischen Folgen eines THC-Konsums zum Ziel hatten und Teile der zitierten Dissertation sind. In der ersten Studie wurden spezifische Effekte des frühen Einstiegsalters, von Konsumdauer und Konsumhäufigkeit auf die Funktionen des Arbeitsgedächstnisses erwachsener Cannabis-Konsumenten untersucht, in der zweiten Studie wurden die gleichen Merkmale auf Zusammenhänge mit Veränderungen von Hippocampus und Parahippocampus untersucht, die mit Auffälligkeiten in der Gedächtnis- und Lernleistung einhergehen (Quellen: B. Becker et al., The impact of early-onset cannabis use of funcitional brain correlates of working memory, Progress in Neuro-Psychopharmacology & Biological Psychiatry, U. K., Elsevier B.V., Amsterdam/Niederlande 2010 und B. Becker et al., Altered parahippocampal functioning in cannabis users is related to the frequency of use, Psychopharmacology, 209, 3/2010, S. 361‑ 374, Springer International Publishing AG, Cham/Schweiz 2010, beide Studiendokumente befinden sich in der Anlage des Pdfs, http://docserv.uni‑duesseldorf.de/...).
In der Einleitung zu ihrer ersten Studie über potentielle Zusammenhänge mit den Leistungen des Arbeitsgedächnis verweisen Becker und sein Team zunächst auf frühere Arbeiten anderer Forscher, die hier einen Zusammenhang nahelegen: „Studien zu kognitiven Langzeiteffekten des Cannabis-Konsums berichten von spezifischen Defiziten in den Bereichen Arbeitsgedächtnis, assoziatives Lernen, Aufmerksamkeit und exekutive Funktionen (Bolla et al., 2002, Solowij et al., 2002b, Messinis et al., 2006). Ergebnisse einiger neurokognitiver Studien legen nahe, dass die Entwicklung und Persistenz dieser Leistungsdefizite vor allem bei Konsumenten auftreten, welche den Konsum im frühen und mittleren Jugendalter beginnen (Ehrenreich et al., 1999, Pope et al., 2003). Während der Adoleszenz führen progressive und regressive Veränderungen an den Nervenverbindungen im Gehirn zu einer Optimierung der kognitiven Leistungsfähigkeit (Spear, 2000, Gogtay et al., 2004, Sowell et al., 2004). Da das endocannabinoide System während dieser Periode fundamentale Prozesse der Gehirnreifung reguliert (Harkany et al., 2007), ist es denkbar, dass die Zufuhr exogener Cannabinoide durch den Konsum von Cannabis in dieser Periode zu Störungen in der normalen Gehirnentwicklung führen könnte und das jugendliche Gehirn während dieser Phase besonders vulnerabel für mögliche negative Folgen des Cannabis-Konsums ist.“ (Quelle: wie oben).
Becker und sein Team untersuchten zwei Gruppen von Cannabiskonsumenten. Die erste Gruppe bestand aus 26 Erwachsenen, die vor ihrem 16. Lebensjahr mit dem Konsum begonnen hatten, die Vergleichsgruppe bestand aus 17 Erwachsenen mit einem späteren Einstiegsalter. Alle mussten Aufgaben lösen, bei denen das Arbeitsgedächtnis involviert war. Die Teilnehmer zeigten bei vergleichbarer Leistung unterschiedlich starke Hirnaktivierungen. Die Konsumenten mit frühem Einstiegsalter wiesen bei vergleichbarer Leistung eine stärkere neuronale Aktivierung der für das Arbeitsgedächtnis zuständigen Hirnareale als auch im superioren Parietalcortex auf. Es wurden auch andere Einflussfaktoren untersucht, zum Beispiel die kumulativ konsumierte Gesamtmenge von Cannabis, die Konsumhäufigkeit zum Zeitpunkt der Studie oder die Abstinenzzeit, jedoch waren bei allen restlichen potentiellen Einflüssen keine Zusammhänge mit erhöhten neuronalen Aktivierungen nachweisbar.
Fazit: „Die Ergebnisse dieser Studie weisen darauf hin, dass das Gehirn während der Adoleszenz besonders vulnerabel für Auswirkungen des Cannabis-Konsums ist. Da die Konsumenten mit einem frühen Einstiegsalter erhöhte neuronale Aktivierung bei gleicher Leistung zeigten, können die Ergebnisse im Sinne einer suboptimalen kognitiven Effizienz als Folge des frühen Einstiegsalters gedeutet werden. Allerdings konnten alternative Erklärungen, wie etwa die Möglichkeit, dass die festgestellten Unterschiede bereits vor dem Konsum bestanden, anhand der vorliegenden Daten nicht ausgeschlossen werden.“ (Quelle: wie oben).
Es soll an dieser Stelle auf die Beschreibungen der Auswirkungen psychosozialen Disstresses auf die Hirnentwicklung Jugendlicher verwiesen werden, insbesondere auf die während der Hirnentwicklungsphasen zwischen Pubertät und dem 25. Lebensjahr (→ Abschnitt 4.12.4 und insbesondere Abbildung 40). Die Graphik ermöglicht es, potentielle Folgen negativer exogener Auswirkungen ‑ zu denen auch Cannabiskonsum gehört ‑ auf das sich entwicklende Gehirn in diesem Zeitraum abzuleiten.
In der zweiten Studie untersuchte Becker mit einem Team nochmal alle drei Merkmale (Einstiegsalter, Konsumdauer und aktuelle Konsummenge) auf Zusammenhänge mit der neuronalen Aktivierung von Hippocampus und Gyrus parahippocampalis, während die Probanden Lern- und Gedächtnisaufgaben bewältigen mussten.
Auch hier verweisen die Autoren in ihrer Studiendokumentation zunächst auf verschiedenen frühere Untersuchungen anderer Forschergruppen, die Zusammenhänge zwischen funktionellen Auffälligkeiten im Hippocampus und Gyrus parahippocampalis während der Lösung von Lern- und Gedächtnisaufgaben bei allen drei Merkmalen des Cannabiskonsums feststellten und leiten ihre eigenen Studienziele ab: „Neurokognitive Studien berichten einen Zusammenhang zwischen kognitiven Leistungsdefiziten bei den Konsumenten von Cannabis und einem jüngeren Einstiegsalter (Ehrenreich et al., 1999, Pope et al., 2003), einer höheren Konsumhäufigkeit (Pope and Yurgelun-Todd, 1996, Pope et al., 2001b) und einer längeren Konsumdauer (Solowij et al., 2002b, Messinis et al., 2006). Nachdem in der ersten Studie ein Zusammenhang mit dem Einstiegsalter und arbeitsgedächtnisassoziierter neuronaler Aktivierung nachgewiesen werden konnte, sollte in der zweiten hier vorgelegten Studie untersucht werden, ob die individuellen Cannabis-Konsumparameter (1) Einstiegsalter, (2) aktuelle Häufigkeit des Konsums und (3) Dauer des regelmäßigen Konsums spezifische Auswirkungen auf die bei Cannabis-Konsumenten berichteten Auffälligkeiten in der gedächtnisassoziierten hippokampalen Aktivierung (Jager et al., 2007b, Nestor et al., 2008) haben. Die Klärung möglicher Zusammenhänge könnte es ermöglichen, besonders riskante Konsummuster und somit Populationen mit einem hohen Risiko für kognitive Folgewirkungen des Cannabis-Konsums zu identifizieren. Darüber hinaus können Zusammenhänge mit spezifischen Parametern des Konsums wichtige Informationen über die Persistenz der Folgewirkungen beinhalten. So schlagen bspw. Solowij und Battisti in ihrem aktuellen Review über die Folgewirkungen des Cannabis-Konsums auf Gedächtnisleistung (Solowij and Battisti, 2008) vor, Zusammenhänge mit der aktuellen Häufigkeit des Konsums als eher subakute und somit reversible Auffälligkeiten zu interpretieren. Im Gegensatz dazu schlagen die Autoren vor, Zusammenhänge mit der Dauer des Konsums und der konsumierten Gesamtmenge als eher langandauernde und somit persistente Auffälligkeiten zu interpretieren.“ (Quelle: wie oben).
Becker und sein Team untersuchten 42 Cannabis-User mit sehr unterschiedlichen Konsummustern hinsichtlich Konsumdauer, Konsumhäufigkeit und Einstiegsalter, die mit einer statistischen Größe (Mediansplit) jeweils pro Merkmal in zwei Grupen unterteilt wurden, nämlich längere oder kürzere Konsumdauer, höhere oder geringere Konsumhäufigkeit und frühes oder spätes Einstiegsalter. Dann wurden die Lernleistungen aller Teilnehmer mit der jeweiligen neuronalen Aktivierung der zu untersuchenden Hirnregionen verglichen. Lediglich die Gruppe mit den verschiedenen Konsumhäufigkeiten zeigte Abweichungen, wobei häufiger Cannabiskonsum zu einer stärkeren paraphippocampalen Aktivierung führte. Auch nach einer Überpüfung der Ergebnisse mithilfe einer statistischen Berechnungsmethode (Lineares Regressionsmodell), bei der die drei Konsummerkmale nochmals mit hippocampalen bzw. parahippocampalen Veränderungen während der Aufgabenlösung verglichen wurden, änderte das am Resultat nichts: „Die Ergebnisse dieser Analyse bestätigten den spezifischen Zusammenhang zwischen einer höheren Konsumhäufigkeit und erhöhter parahippokampaler Aktiviät, auch unter Berücksichtigung des Einstiegsalters und der Konsumdauer.“ (Quelle: wie oben). Die Studienverantwortlichen deuten die Ergebnisse als Hinweise auch auf reversible Effekte des Cannabiskonsums bezogen auf den Gyrus parahippocampalis.
Abgesehen davon, dass beide Studien aufgrund anderer Zielrichtungen keine Aussagen hinsichtlich eines Zusammenhangs zwischen Cannabiskonsum und dem Auftreten persistenter Affektstörungen machen, wurden sie in den Jahren 2009 bis 2010 durchgeführt, so dass neuere Erkenntnisse einer sich rasant entwickelnden Forschung nicht berücksichtigit werden konnten. Die relativ geringe Teilnehmerzahl beider Studien ist kritikwürdig.
Dennoch ergeben sich erste wertvolle Hinweise, auch wenn die Studienergebnisse Arbeitsgedächtnis, Lernfähigkeit und auffällige Aktivitäten im Gyrus parahippocampalis betrafen:
- Ein früher Cannabiskonsum in der Jugend wirkt sich vermutlich langfristig negativ auf die Gehirnleistungsfähigkeit aus.
- Ein häufiger Cannabiskonsum wirkt sich vermutlich eher reversibel kurzfristig negativ auf die Gehirnleistungsfähigkeit und bestimmte Hirnareale auf, so dass nach einer Cannabisabstinenz die ursprüngliche Hirnleistungsfähigkeit wiederhergestellt ist.
Interessant und hilfreich ist die schon kurz erwähnte und relativ aktuelle Übersichtsarbeit eines Teams um Eva Hoch aus dem Jahre 2015, die im Deutschen Ärzteblatt erschien, und an der Fachärzte aus verschiedenen deutschen Universitäten bzw. Universitätsklinken oder Instituten ‑ Heidelberg, Mannheim, Duisburg/Essen, Göttingen und Halle-Wittenberg ‑ beteiligt waren. In der Arbeit werden Studien zitiert, bei denen Zusammenhänge zwischen Cannabis und verschiedenen kognitiven und psychiatrischen Zuständen untersucht wurden (Quelle: E. Hoch, R. Thomasius, F. Ganzer et al., Risiken bei nichtmedizinischem Gebrauch von Cannabis, Deutsches Ärzteblatt, 112 (16), erschienen am 17.4.2015, Deutscher Ärzteverlag GmbH, Berlin 2015, https://www.aerzteblatt.de/...). Die Autoren haben bei einigen Arbeiten deren Evidenzgrade nach CEBM angegeben (→ Glossar über Evidenzkriterien).
TABELLE 25: KOGNITIVE UND PSYCHIATRISCHE AUFFÄLLIGKEITEN BEI CANNABISKONSUM
Studie |
Kognitive Auffälligkeiten bzw. psychiatrische Komorbiditäten |
|---|---|
| I. Grant, T. Wolfson et al., Non-acute (residual) neurocognitive effects of cannabis use: A meta- analytic study, Journal of the International Neuropsychological Society, Vol. 9 (5), 7/2003, Evidenzgrad 1a, https://www.cambridge.org/... |
Die Meta-Studie schloss elf Studien mit insgesamt 623 Cannabiskonsumenten und 409 Nichtkonsumenten bzw. Minimal-Usern ein. Leicht ungünstige Effekte auf Lernvermögen und Gedächtnis bei gewohnheitsmäßigem Cannabiskonsum, die auch noch nach einem mindestens 24stündigen Konsumstopp nachweisbar waren. Keine Beeinträchtigungen bei Aufmerksamkeit und Reaktionsgeschwindigkeit. |
| A. M. Schreiner, M. E. Dunn, Residual Effects of Cannabis Use on Neurocognitive Performance After Prolonged Abstinence: A Meta-Analysis, Experimental and Clinical Psychopharmacolgy, Online-Publikation 6/2012, Evidenzgrad 1a, Pdf unter https://www.researchgate.net/... oder https://www.ncbi.nlm.nih.gov/pubmed/... |
Die Autoren beziehen sich auf zwei Meta-Studien, von denen sie eine ältere aktualisierten (insgesamt 33 Studien) und eine zweite (mit 13 Studien) selber erstellten. Es wurden jeweils dauerhafte Symptomatiken nach einer Abstinenzzeit von mindestens 25 Tagen festgestellt und bewertet. Bei akutem Konsum stellte man eine leicht reduzierte allgemeine kognitive Leistungsfähigkeit im Vergleich mit Nicht-Konsumenten fest; diese betraf abstraktes Denken einschließlich exekutiver Hirnleistungen, Lernen, Aufmerksamkeit, Merkfähigkeit und Psychomotorik. Nach mindestens 25tägigem Konsumstopp waren diese Symptome nicht mehr nachweisbar. |
| K. L. Medina, S. Tapert et al., Neuropsychological functioning in adolescent marijuana users: Subtle deficits detectable after a month of abstinence, Journal of the International Neuropsychological Society, Vol. 13 (5), 9/2007, https://www.ncbi.nlm.nih.gov/... N. Solowij, K. A. Jones et al., Verbal learning and memory in adolescent cannabis users, alcohol users and non-users, Psychopharmacology 2011, Vol. 216, Pdf unter http://link.springer.com/... |
Im Gegensatz zu den beiden Meta-Studien von Schreiner und Dunn ergaben diese beiden Untersuchungen, dass bei frühem Cannabiskonsum schon in der Jugend kognitive Einschränkungen auch nach vierwöchiger Abstinenz noch feststellbar waren. Es zeigten sich leichte bis mittlere Einschränkungen in der Psychomotorik und bei kognitiven Funktionen. Betroffen waren psychomotorische Geschwindigkeit, Aufmerksamkeit, Planungsfähigkeit und das Gedächtnis. |
| M. H. Meier, A. Caspi, T. E. Moffitt et al., Persistent cannabis users show neuropsychological decline from childhood to midlife, Proceedings of the National Academy of Sciences of the United States of America (PNAS), Online-Publikation am 27.8.2012, Evidenzgrad 1b, http://www.pnas.org./... |
Die Auswertung einer früheren prospektiven Studie mit 1.037 Teilnehmern aus der neuseeländischen Otago-Dunedin-Region (bekannt als „Dunedin-Studie“) mit Teilnehmern der Geburtsjahrgänge 1972 und 1973, die bis zum 38. Lebensjahr begleitet wurden, mit einer neurologisch-psychologischen Untersuchung im 13. Lebensjahr und fünf Erhebungen (18., 21., 26., 32. und 38. Lebensjahr) bezüglich des Cannabiskonsums ergab einen negativen Einfluss regelmäßigen Cannabiskonsums in der Jugend auf die spätere Intelligenz. Die Untersuchung verglich die Intelligenzleistung früher Cannabis-User (vor Eintreten ihrer Volljährigkeit) mit solchen, die erst als Erwachsene damit anfingen. Bei den Teilnehmern der ersten Gruppe war über die Jahre ein Rückgang der Qualität neuropsychologischer Funktionen feststellbar. Sie hatten im Alter von 38 Jahren einen im Schnitt um acht Punkte geringeren Intelligenzquotienten als mit 13 Jahren. Auch eine Beendigung des Konsums konnte die neuropsychologischen Probleme nicht vollständig beseitigen. In der zweiten Gruppe war kein derartiger Rückgang festzustellen. In der Arbeit wurden Störeffekte ausgeschlossen, beispielsweise akute Cannabisvergiftungen, Konsum anderer Substanzen, unterschiedliche Qualiäten der Ausbildung der Teilnehmer oder eine Erkrankung an Schizophrenie. Die Autoren bewerten ihre Ergebnisse so: „Die Befunde lassen auf eine neurotoxische Wirkung von Cannabis auf das jugendliche Gehirn schließen und machen deutlich, wie wichtig die Prävention für Jugendliche und entsprechende politische Maßnahmen zur Aufklärung sind.“ |
| K. M. Lisdahl, S. Shollenbarger et al., Dare to Delay? The Impacts of Adolsescent Alcohol and Marijuana Use Onset on Cognition, Brain Structure, and Function, Front Psychiatry, Online- Publikation am 15.4.2013, Evidenzgrad 2a, https://www.ncbi.nlm.nih.gov/... |
In ihrer Einleitung verweisen die Autoren dieser Überblicksstudie auf Untersuchungen, die signifikante langfristige neurodegenerative Veränderungen von grauer und weißer Hirnsubstanz im Zusammenhang mit Alkohol- und Marihuanakonsum Jugendlicher und junger Erwachsener feststellten (Giedd et al. 1996, Paus et al. 1999, Sowell et al. 1999, 2002 und 2004, Gogtay et al. 2004, Barnea-Goraly et al. 2005, Lenroot und Giedd 2006). Auch in mehreren Tierstudien wurden negative Wirkungen beider Substanzen auf das sich noch entwickelnde Nervensystem festgestellt. In ihrer Arbeit analysierten die Autoren mehrere Studien, bei denen die Wirkungen von Alkohol und Marihuana im frühen Einstiegsalter auf neurokognitive Funktionen untersucht wurden, u. a. schloss diese auch die schon beschriebene Studie von M. H. Meier et al. mit ein. In der Zusammenfassung weisen sie darauf hin, dass es evidente Hinweise auf eine massive langfristige Schädigung von Kognition, Hirnstruktur und Hirnfunktionalität durch einen Konsumbeginn Jugendlicher und junger Erwachsener gibt, dieses Wissen aber sehr gut für medizinische Programme und zielgruppenspezifische Aufklärung nutzbar sei und um Präventionsmaßnahmen zu initiieren. |
| J. Cousijn, L. Koenders et al., Relationship between working-memory network function and substance use: a 3-year longitudinal fMRI study in heavy cannabis users and controls, Addiction Biology, Vol. 19 (2), Online-Text vom 25.11.2013, Pdf unter https://www.researchgate.net/... oder http://onlinelibrary.wiley.com/... |
Bei erwachsenen Cannabis-Usern, die schon als Jugendliche oder junge Erwachsene konsumierten, wurden keine Beeinträchtigungen des Arbeitsgedächtnisses festgestellt, ebenfalls keine Veränderungen der Hirnaktivitäten während der Tests. Die Kritik an der Studie bezieht sich auf die geringe Anzahl der Teilnehmer (49), die als zu einfach bewerteten Testaufgaben und die mangelnde Vergleichbarkeit der jeweiligen Einstiegsalter der Teilnehmer, die nur eingeschränkt als früh bezeichnet werden können. |
| J. Cousijn, A. E. Goudriaan et al., Grey matter alterations associated with cannabis use: Results of a VBM study in heavy cannabis users and healthy controls, NeuroImage, 59 (4), 2012, http://www.sciencedirect.com/... |
Cousijn und ihr Team untersuchten Veränderungen der Hirnmorphologie in Arealen mit besonders vielen CB1-Rezeptoren (orbifrontaler Cortex, Gyrus cinguli anterior, Striatum, Amygdala, Hippocampus, Kleinhirn) bei 33 aktiven Langzeitkonsumenten (sogenannte „Heavy‑User“) ausschließlich von Cannabis und verglichen die Ergebnisse mit einer gesunden Kontrollgruppe von 42 Personen. Es zeigten sich Veränderungen an der grauen Substanz (= Neuronen) in der Amygdala und im Hippocampus. Auch korrelierte die Abnahme des Volumens mit der Menge des Cannabisgebrauchs. In einem Bereich des Kleinhirns ergab sich eine Volumenvergrößerung bei Cannabiskonsumenten. Keine Veränderungen wurden beispielsweise in der weißen Substanz, den Nervenfasern, festgestellt. |
| A. Zalesky et al., Effect of long-term cannabis use on axonal fibre connectivity, Brain, Vol. 135 (7), 7/2012, Evidenzgrad 1b, https://academic.oup.com/... |
In dieser Studie wurden 59 „Heavy‑User“ von Cannabis mit 33 Nicht-Konsumenten verglichen. Es gibt es Hinweise auf mikrostrukturelle Veränderungen axonaler Faserwege der weißen Hirnsubstanz in Zusammenhang mit einem früh begonnenen Langzeitkonsum von Cannabis. Die Schädigungen korrelierten sehr deutlich mit dem Einstiegsalter und wurden mit verschiedenen Dichtemessungen mittels einer Diffusions-Tensor-Bildgebung (DW‑MRT) festgestellt. Sie betrafen den rechten Fornix (Fimbrien), ein bestimmtes Axonbündel vom Splenium des Corpus Callosum zum rechten Precuneus ‑ hier sogar ein Rückgang um 88% ‑ und Faserbündel zwischen beiden Hirnhälften (Commisualfasern). Die Autoren kommen zu dem Schluss, dass ein längerfristiger Konsum von Cannabis für die weiße Substanz eines sich entwickelnden Gehirns gefährlich ist. Je später der Einstieg in den Konsum, desto weniger ausgeprägt waren die Beeinträchtigungen. |
| L. Horwood, D. Fergusson et al., Cannabis use and educational achievement: Findings from three Australian cohort studies, Drug and Alcohol Dependence, 110/2010, Evidenzgrad 1a, http://www.sciencedirect.com/science/ |
Mit dieser australischen Kohortenstudie über mehr als 6.000 Teilnehmer wurden Zusammenhänge zwischen der schulischen Leistung hinsichtlich Schulabbruch und Bildungsniveau (High School, Universität, Abschlussgrad) und dem Beginn eines Konsums von Cannabis untersucht (vor dem 15. Lebensjahr, zwischen dem 15. und 17. Lebensjahr, nach dem 18. Lebensjahr). Es wurden robuste Zusammenhänge zwischen den Merkmalen festgestellt. So war ein früher Cannabiskonsum vor dem 15. Lebensjahr deutlich mit einem Schulabbruch bzw. einem verminderten Bildungsniveau assoziiert. |
| R. C. Kessler, H. Zheng et al., The US National Comorbidity Survey Replication (NCS-R): design and field procedures, International Journal of Methods in Psychiatric Research, 2004, Vol. 13 (2), https://ai2-s2-pdfs.s3.amazonaws.com/... |
Bei 50 bis 90% aller abhängigen Cannabiskonsumenten wird mindestens einmal in ihrem Leben (Lebenszeit-Prävalenz) eine weitere psychiatrische Störung, einschließlich des Gebrauchs von Alkohol und anderen Rauschsubstanzen, diagnostiziert. |
| M. Gibbs et al., Cannabis use and mania symptoms: A systematic review and meta- analysis, Journal of Affective Disorders, Vol. 171, 15.1.2015, http://www.sciencedirect.com/... |
Die Literaturstudie schloss sechs Arbeiten mit insgesamt 2.391 Personen ein, die Symptome einer Manie gehabt hatten bzw. hatten. In allen Arbeiten wurde ein Zusammenhang zwischen Cannabiskonsum und einer manisch-depressiven Erkrankung festgestellt: Die Manie verschlechterte sich bei konsumierenden Teilnehmern, die zuvor als manisch-depressiv diagnostiziert wurden. |
| S. M. Strakowski, M. P. DelBello, J. Amicone et al., Effects of Co-occurring Cannabis Use Disorders on the Course auf Bipolar Disorder After a First Hospitalization for Mania, Archives of General Psychiatry (JAMA Psychiatry), Vol. 64, 1/2007, http://jamanetwork.com/... |
Von den 144 Teilnehmern mit einer Bipolaren Störung Typ 1 (schnelle Wechsel zwischen Manie und Depression bzw. gemischte Phasen) ging bei 33 Teilnehmern ein Cannabismissbrauch der Erkrankung voraus bzw. bei 36 folgte der Cannabismissbrauch der Erkrankung, bei 75 Teilnehmern lag kein Missbrauch vor. Die Teilnehmer der ersten Gruppe (Cannabismissbrauch zuerst) erholten sich während eines Entzugs und Krankenhausaufenthalts besser als die zweite Gruppe. Cannabis war allgemein mit häufigeren akuten Erkrankungsepisoden assoziiert, nach der Behandlung war die Rückfallqoute generell hoch. Diese konnte reduziert werden, wenn sofort nach dem ersten Klinikaufenthalt eine Suchttherapie durchgeführt wurde. |
| H. M. X. Lai, T. Sitharthan, Exploration of the Comorbidity of Cannabis Use Disorders and Mental Health Disorders among Inpatients Presenting to All Hospitals in New South Wales, Australia, The American Journal of Drug and Alcohol Abuse, Vol. 38 (6), 2012, http://www.tandfonline.com/... |
In die umfangreiche Auswertung einbezogen waren etwa 1,8 Millionen Patienten in stationärer Behandlung zwischen Juli 2006 und Juni 2007 im australischen Bundesstaat New South Wales. 53,8% der Cannabis konsumierenden Patienten hatten mindestens eine diagnostizierte psychische Störung, insbesondere bei Frauen und Personen zwischen dem 30. und 49. Lebensjahr wurde eine hohe Komorbiditätsrate festgestellt. Die am häufigsten beobachteten psychiatrischen Erkrankungen waren Schizophrenie (15%), Major Depression (10,9%), Persönlichkeitsstörung (9,2%), Belastungsstörung (8,7%), Bipolare Störung (5,7%) und Angststörung (3,4%). |
| L. R. Kvitland, I. Melle, P. A. Ringen et al., Cannabis use in first-treatment bipolar I disorder relations to clinical characteristics, Early Intervention in Psychiatry, Vol. 10 (1), 2/2016, http://onlinelibray.wiley.com/... |
101 Patienten, die sich erstmals wegen ihrer Bipolaren Störung Typ 1 (schnelle Wechsel zwischen Manie und Depression und gemischte Phasen) behandeln ließen, wurden hinsichtlich ihres Cannabiskonsums innerhalb der letzten sechs Monate vor Studienbeginn, ihrer akutellen manischen, depressiven oder psychotischen Symptomatik und des Alters bei Beginn des ersten Krankheitsschubs analysiert. Patienten, deren letzter Cannabiskonsum zeitlich nahe dem Behandlungsbeginn lag, waren beim ersten manischen oder psychotischen Krankheitsschub deutlich jünger als der Rest der Teilnehmer und unternahmen häufiger einen Selbstmordversuch. Das Studienteam sieht bestätigt, dass kürzlicher Cannabiskonsum mit einem niedrigeren Alter bei Ausbruch der Erkrankung einhergeht und auch mit einer höheren Suizidversuchsrate verbunden ist. Sie schließen daraus, dass kürzlicher Konsum mit einem schwereren Krankheitsverlauf in der Beginnphase assoziiert. |
| C. Silberberg et al., Cannabis, cannabinoids and bipolar disorder, in: Marijuana and Madness, Chapter 11, 2nd edition, Cambridge University Press, 2012, https://books.google.de/... |
Die Autoren dieser Literaturstudie zitieren die Ergebnisse zahlreicher Untersuchungen des Zusammenhangs zwischen Cannabis und einer manisch-depressiven Erkrankung. Ihr Resümee entspicht den Ergebnissen einiger der vorher zitierten Studien, beispielsweise von Kvitland et al.: „Es gibt keinen Zweifel an der hohen Komorbidität zwischen einer Bipolaren Störung und Cannabis. Es mehren sich auch die Nachweise der Schädlichkeit seines Gebrauchs bei Patienten mit einer Bipolaren Störung. Cannabiskonsum ist nicht nur mit höheren Risiken für den Ausbruch manischer Phasen verbunden, Studien legen nahe, dass der Beginn einer Erkrankung bei Cannabis-Usern früher eintritt. Ebenfalls senkt Cannabiskonsum die positiven Wrkungen therapeutischer Maßnahmen gegen Bipolare Störungen. Patienten mit einer Bipolaren Störungen haben höhere kognitive Defizite in Bezug auf exekutive Funktionen. Es gibt Hinweise auf neurostrukturelle Unterschiede, wenn auch mit geringer Evidenz und erste Hinweise, dass das endocannabinoide Transmittersystem in pathophysiologische Prozesse der Bipolaren Störung involviert ist. (...)“. |
| C.Henquet, L. Krabbanedam et al., Cannabis use and expression of mania in the general population, Journal of Affective Disorders, Vol. 95 (1 - 3), 10/2006, http://www.jad-journal.com/... |
Die Studie belegt einen positiven Zusammenhang von Cannabiskonsum und vermehrt auftretenden manischen Symptomen bei bipolaren Erkrankungen. |
| S. Lev-Ran et al., The association between cannabis use and depression: a systematic review and meta-analysis of longitudinal studies, Psychological Medicine, Vol. 44 (4), 3/2014, Evidenzgrad 2a, https://www.cambridge.org/... |
Es wurden 57 Studienreports analysiert, die sich mit Zusammenhängen zwischen Cannabiskonsum und der Entstehung bzw. dem Verlauf einer unipolarer Depression beschäftigten; davon wurden 14 Arbeiten exakt quantitativ analysiert und verglichen (mit insgesamt 76.058 Individuen). Das Quotenverhältnis (Odds Ratio) für durchschnittliche Cannabiskonsumenten mit einer Depression betrug 1,17 im Vergleich mit einer Kontrollgruppe und das für „Heavy-User“ mit frühem Beginn und starkem Konsum betrug dagegen 1,62. Beides weist auf positive Zusammenhänge zwischen den Merkmalen hin, insbesondere bei „Heavy-Usern“. |
| E. Manrique-Garcia et al., Cannabis use and depression: a longitudinal study of a national cohort of Swedish conscripts, BMC Psychiatry, 12/2012, https://bmcpsychiatry.biomedical.com/... |
Nach Ausschaltung aller Störfaktoren ergab diese schwedische Kohortenstudie keinen Zusammenhang zwischen Cannabiskonsum und einer Depression bzw. Suizid im Erwachsenenalter, wobei das Alter der Cannabiskonsumenten zwischen 18 und 20 Jahren lag, so dass sich hier keine Aussagen über die Folgen eines früheren Beginns ableiten lassen. Dagegen wurde eine hohe Assoziation zwischen Cannabiskonsum und Schizoaffektiven Störungen festgestellt. |
| W. Pedersen, Does cannabis use lead to depression and suicidal behaviours? A population-based longitudinal study, Acta Psychiatrica Scandinavica, Vol. 118 (5), 11/2008, Evidenzgrad 3a, http://onlinelibrary.wiley.com/... |
Die Studie aus Norwegen kann keinen Nachweis für einen Zusammenhang zwischen Cannabiskonum und Depression erbringen, jedoch wurden verstärkt Suizidgedanken bei Cannabis konsumierenden Jugendlichen und jungen Erwachsenen beobachtet. |
| W. Hall, L. Degenhardt, The adverse health effects of chronic cannabis use, Drug Testing and Analysis, Vol. 6 (1/2), 1/2014, http://onlinelibrary.wiley.com/... |
In diesem Vergleich wurden verschiedene Studien über Cannabiswirkungen analysiert, u. a. auch solche zu den Merkmalen Depression, Bipolare Störung und Suizid. Bei der Depression waren die Ergebnisse uneinheitlich und eher schwach, bei der Bipolaren Störung deuten einige Ergebnisse auf einen Kausalzusammenhang hin, ohne jedoch eindeutig zu sein. Beim Suizid waren die Ergebnisse ebenfalls heterogen, und es gab weder eine eindeutige Aussage über die Höhe des Suizidrisikos noch über die Kausalität. |
| T. H. Moore, S. Zammit. A. Lingford-Hughes et al., Cannabis use and risk of psychotic or affecitve mental health outcomes: a systematic review, The Lancet, Vol. 370 (9584), July/August 2007, http://www.sciencedirect.com/... |
Es wurden 35 Studien verschiedener Publikationen des Zeitraums September 2006 auf Erkenntnisse über langfristige Folgen des Cannabiskonsums hinsichtlich psychiatrischer Erkrankungen analysiert. Es wurde ein erhöhtes Psychoserisiko für alle Personen festgestellt, die jemals Cannabis konsumierten (Odds Ratio 1,41), wobei das Risiko bei häufigem Gebrauch ansteigt (Odds Ratio 2,09). Die Ergebnisse bezüglich Affektiver Störungen, Suizid und Angstzustände war weniger konsistent. |
| S. Lev-Ran, B. Le Foll, K. McKenzie et al., Bipolar disorder and co-occurring cannabis use disorders: Characteristics, co-morbidities and clinical correlates, Psychiatry Research, 209 (3), 10/2013, http://www.sciencedirect.com/... |
Unter einer Stichprobe von 1.905 Individuen mit lebenslanger Prävalenz für eine bipolare Störung betrug der Anteil der Cannabiskonsumenten 7,2% gegenüber 1,2% der Vergleichsgruppe. Bei bipolaren Cannabiskonsumenten ist die Erkrankung mit einem schlechteren Verlauf, das heißt einer größeren Anzahl bipolarer Episoden, und einem früheren Beginn assoziiert. Gleichzeitig hatten diese Personen ein erhöhtes Risiko für weitere Nikotin-, Alkohol- und Drogenkonsumstörungen. |
| L. Degenhardt, C. Coffey, H. Romaniuk et al., The persistence of the association between adolescent cannbis use and common mental disorders into young adulthood, Addiction, 108 (1), 1/2013, Evidenzgrad 2b, https://www.ncbi.nlm.nih.gov/... |
Hohe Assoziationen zwischen Cannabisgebrauch und Angsterkrankungen wurden in dieser australischen Studie ermittelt: Täglicher Konsum in der Jugend ging mit einer Angststörung im Alter von 29 Jahren einher (Odds Ratio 2,5), bei Teilnehmern, die ihren Konsum auch noch im 29. Lebensjahr täglich fortsetzen, betrug diese sogar 3,2. |
| F. S. Stinson, W. J. Ruan et al., Cannabis use disorders in the USA: prevalence, correlates and co-morbidity, Psychological Medicine, 36 (10), 10/2006, Pdf unter https://www.cambridge.org/... |
Die Daten einer großen nationalen repräsentativen Umfrage in den USA mit 43.093 Erwachsenen wurden hinsichtlich verschiedener Assoziationen zwischen Cannabismissbrauch bzw. Cannabisabhängigkeit und einer Vielzahl psychiatrischer Erkrankungen untersucht. Die Ergebnistabelle befindet sich auf Seite 1.454 oben im verlinkten Pdf. Es wurden überall erhöhte Werte gefunden, die Ergebnisse sprechen für sich. Insbesondere Angst- und Persönlichkeitsstörungen wiesen hohe Odds-Ratio-Werte auf, aber auch bei Affektstörungen lagen die Werte i. d. R. zwischen knapp unter 2,0 und knapp über 7,0. |
Tabelle 25: Zusammenfassende Übersicht über einige Ergebnisse der Literaturstudie von E. Hoch, R. Thomasius, F. Ganzer et al., Quelle wie oben. Die Angaben der Evidenzgrade beziehen sich auf die Skala des CEBM (Oxford Centre for Evidence-based Medicine).
(wird fortgesetzt)
3,4-Methylendioxyd-N-methylamphetamin (MDMA, Ecstasy)
(Text folgt)
Methamphetamin (Pervitin, Crank, Crystal Meth)
(Text folgt)
Kokain
(Text folgt)
Diamorphin (Heroin)
(Text folgt)
Halluzionogene
(Text folgt)
4.14 Biotische biologisch-medizinische Noxen ▲
Zur Systematik der Darstellungen biotischer biologisch-medizinischer Noxen
(Text folgt)
Körpertemperatur (Hitzeschockproteine)
(Text folgt)
Aspekte der Blut-Hirn-Schranke
(Text folgt)
Schlafstörungen
(Text folgt)
Infektionserkrankungen
(Text folgt)
Chronisch-entzündliche Erkrankungen
(Text folgt)
Schilddrüsenunterfunktion
(Text folgt)
Vaskuläre Erkrankungen
(Text folgt)
Weitere Erkrankungen des Zentralnervensystems und ihr Einfluss auf Affekte bzw. Affektstörungen
(Text folgt)
Die Durchführung medizinischer Eingriffe unter dem Einfluss narkotisierender Substanzen
(Text folgt)
4.15 Exkurs: Gibt es direkte Abhängigkeiten zwischen Symptomen?▲
Mit Abschnitt 4.14 endet die systematische Analyse potentieller Auswirkungen verschiedener exogener Noxen auf das Zentralnervensystem im Hinblick auf Affektive Störungen aus einer kausaltheoretischen, ganzheitlichen Perspektive.
Zum Abschluss soll nun die Frage in den Mittelpunkt rücken, welche Ansätze die Modelle zur Erklärung der auf den ersten Blick unübersichtlichen Beziehungen zwischen Symptomen Affektiver Störungen bieten ‑ insbesondere, ob Symptome als Noxen weitere Symptome auslösen können. Denn in den meisten Fällen müssen depressiv Erkrankte nicht nur gegen den Stimmungsabfall als Hauptsymptom kämpfen, sondern auch gegen weitere Symptome, zum Beispiel Antriebsstörungen, die häufig zum vollständigen Verlust des Interesses an Aktivitäten führen (→ Abschnitt 1.1). Kausaltheoretisch wird zwischen multifaktoriellen Primärauslösern, Ursachen auf der hirnorganischen Ebene und Erkrankungssymptomen jedenfalls klar differenziert.
Verursacher der Erkrankung und ihrer verschiedenen Symptome sind aus dieser Sicht entweder Kausalfaktormängel (→ Kapitel 4, Teil A) und/oder exogene Noxen (→ Kapitel 4, Teil B), die zu Problemen der Reizverarbeitung führen, da sie funktional-strukturelle Veränderungen in genau den Arealen des Zentralnervensytems bewirken, die mit der Entstehung und Steuerung von Affekten in Verbindung gebracht werden.
Damit sind direkte Kausalbeziehungen zwischen Symptomen prinzipiell ausgeschlosssen. Es muss an dieser Stelle noch einmal darauf hingewiesen werden, dass hier nicht von depressiven Verstimmungen die Rede ist, sondern von einer (klinischen) Erkrankung, so wie im Kapitel 1 beschrieben.
Aber sind Verursacher und Symptome einer (klinischen) Depression in der Realität immer strikt zu trennen? Manchmal scheint es so, als ob Symptome direkt andere Symptome hervorrufen könnten. Konkrete Beispiele: Aus Stimmungstiefs resultieren direkt Antriebs‑ und Interessenverluste oder umgekehrt Antriebs‑ und Interessenverluste wären augenscheinlich in der in der Lage, Stimmungstiefs zu verursachen.
Träfe dies zu, würde die Grenze zwischen Auslöser und Symptom komplett verwischt, was im Gegensatz zu den in kausaltheoretischen Modellen beschriebenen Mechanismen stände. Es bedeutet, sich einer unübersichtlichen Gemengelage zwischen Auslösern und Symptomen gegenüberzustehen, die plausible und eindeutige Erklärungen der Erkrankungsursprünge unmöglich machten.
Gerade in Psychologie und psychotherapeutisch orientierter Psychiatrie werden direkte Ursache‑Wirkungs‑Beziehungen zwischen Symptomen gerne und häufig konstruiert, denn sie bieten einfache Erklärungsmuster komplexer Zusammenhänge und dienen darüber hinaus auch der Begründung simpler (häufig verhaltens‑)therapeutischer Maßnahmen mit dem Anspruch, Affekterkrankungen dadurch langfristig beseitigen zu können. Aber auch solche rein aus psychologischer Perspektive gemachte Annahmen sind physiologisch zu begründen. Es muss schließlich auch eine Erklärung dafür geben, warum bei einem Teil der Erkrankten Stimmungstiefs nicht in Verbindung mit Antriebsproblemen einhergehen und umgekehrt ‑ und dazu wird eine plausible Begründung benötigt.
Kausale Zusammenhänge von Symptomen aus kausaltheoretischer Sicht
Der kausaltheoretische Standpunkt ermöglich nämlich durchaus plausible Erklärungen, wie zum Beispiel Stimmungstiefs langfristig in Antriebsstörungen oder umgekehrt Antriebsstörungen in Stimmungstiefs münden - jedoch nicht direkt, sondern indirekt über den Stressmechansimus.
Stress führt nämlich zu einer übermäßigen Produktion des Nervengifts Cortisol, das funktionale und strukturelle neurologische Schädigungen im Zentralnervensystem zu provozieren vermag. Stress stellt damit eine Noxe dar, die via Cortisol ähnlich der von außen einwirkenden Noxen, beispielsweise Nervengiften oder schädlicher Strahlung, wirkt (→ Abschnitt 4.12). Je nachdem, welche Areale des Gehirns durch Cortisol geschädigt werden, sind unterschiedliche Symptome die Folgen. Annahme ist, dass die Steuerung verschiedener Affekte entweder in unterschiedlichen Regionen innerhalb des affektrelevanten Gesamtareals lokalisiert sind oder sich in unterschiedlichen neuronalen Verschaltungsmustern widerspiegeln:
Symptom 1 → Stress → Cortisol → Schädigung affektrelevanter Hirnareale → Symptom 2
Das zugrundeliegende Ursache-Wirkungs-Muster gilt nicht nur für den Primärauslöser Stress/Cortisol, sondern für sämtliche Primärauslöser, d. h. auch für multiple Veränderungen der ursprünglichen Erbinformation, Genregulationsprobleme durch ncRNA-Mangel, Nervengifte oder ionisierende Strahlung, beispielsweise Radioaktiviät, und für alle anderen hier diskutierten Primärauslöser:
Primärauslöser → Schädigung affektrelevanter Hirnareale → Symptom einer Affekterkrankung
Korrelative Zusammenhänge von Symptomen aus kausaltheoretischer Sicht
Kausalität besteht gemäß den Modellen zwischen einem Primärauslöser und Symptomen in Form einer einfachen, dreistufigen Kette, während die Symptome untereinander immer korrelativ miteinander verbunden sind.
Eine Korrelation zwischen den Symptomen ist mit der organischen Einheit affektverarbeitender Hirnareale zu begründen. Affektstörungen sind symptomatisch durch ein annähernd paralleles Geschehen von Stimmungsabfall, Aktivitätsrückgang, dem Verlust von Interessen und weiteren Symptomen gekennzeichnet. Dass verschiedene Symptome in zeitlichen Abständen auftreten, ist kein Hinweis auf einen Kausalzusammehang.
Ein starkes Argument für diese Sicht ist ‑ neben einer potentiellen Multifunktionalität affektrelevanter Gehirnareale ‑ die Existenz von Nervennetzen, die verschiedene affektive (Sub‑)Systeme miteinander verbinden, wobei hier an erster Stelle die Formatio reticularis zu nennen ist (→ Abschnitt 1.3.1). Selbst eine Störung der Formatio reticularis durch eine Veränderung an nur einer Stelle des Gesamtsysstems könnte aufgrund der komplexen Neuronenverschaltung Auswirkungen auf das gesamte affekverarbeitende System haben und weitere Symptome verantworten.
ABBILDUNG: SYMPTOME AFFEKTIVER STÖRUNGEN UND KORRELATIVITÄT
Abbildung: Ein komplexes Areal des Zentralnervensystems ist für Affekte und Antrieb zuständig und wird durch grüne Flächen symbolisiert. Es gibt in diesem Bereich erhebliche Differenzierungen, denn unterschiedliche Affekte werden auch auf unterschiedliche Weisen verarbeitet bzw. verschiedene Affekte entstehen überhaupt erst durch Unterschiede in der funktionalen und strukturellen Verarbeitung, beispielsweise in räumlich voneinander abgegrenzten Sub-Arealen oder durch verschiedenartige Neuronenverschaltungen oder unterschiedliche Verbindungswege mit anderen großen Hirnarealen, beispielsweise zum frontalen Cortex. Darüber hinaus sind alle affektrelevanten Sub-Areale durch das Nervenetz der aufsteigenden Formatio reticularis komplex untereinander und mit anderen höheren Gehirnregionen verknüpft. Details zu den genauen Mechanismen oder Wegen sind nicht entscheidend, in dieser Zeichnung ist ein Szenario beispielhaft anhand verschiedener Affekte verdeutlicht (Stimmung, Antrieb, Motivation, Interessen). Durch diese komplizierten strukturellen Verbindungen innerhalb des affektrelevanten Gesamtareals kann sich eine pathologische Veränderung an einer Stelle auf alle affektiven Hirnleistungen auswirken. Ebenso können verschiedene Sub-Areale unabhängig voneinander von einem oder auch mehreren Primärauslösern geschädigt werden. Symptome einer Affekterkrankung stehen aus diesen Gründen untereinander in korrelativem Zusammenhang. Primärauslöser der pathologischen hirnorganischen Veränderungen können potentiell sämtliche in Kapitel 4 aufgezählten endogenen und exogenen Noxen sein.
Zum nächsten Kapitel 5 - Kausale Therapien - bitte hier klicken: ►